
タンパク質と小分子(リガンド)がどのように結合するか、または結合しないのか、正確に予測(ドッキング予測)することができれば、様々な応用が可能です。近年、DiffDock1に代表される拡散モデル(diffusion model)ベースの手法がドッキングの速度と精度向上に大きく貢献しましたが、生成されたポーズに原子衝突が含まれていたり、化学的に不自然なねじれ角が生じる「物理的に非現実的な構造」になりやすいという問題があります。今回は、この物理的妥当性の問題を解決しつつ、推論速度も大幅に改善したパイプライン「MATCHA」について勉強しました。
※この記事の作成には、文章の校正の一部で生成AIを活用しています。最終的な内容は、全て筆者が確認・編集しています。
MATCHA 論文:Frolova D. et al. MATCHA: Multi-stage Riemannian Flow Matching for Accurate and Physically Valid Molecular Docking. (2025)arXiv:2510.14586 .doi: 10.48550/arXiv.2510.14586
MATCHA GitHub(リポジトリ):Matcha https://github.com/LigandPro/Matcha
1. 研究の背景と目的
深層学習ベースのドッキングが物理的妥当性を犠牲にしやすい原因は、モデルが「リガンドの幾何学的制約」を暗黙的にしか扱えないことにあります。リガンドの動きには3種類の自由度があり、それぞれが数学的に異なる空間に属します。
- 平行移動(Translation):3次元ユークリッド空間 ℝ³ — タンパク質中心に対するリガンドの重心位置
- 回転(Rotation):特殊直交群 SO(3) — リガンドの全体的な向き
- ねじれ角(Torsion):円周空間 SO(2)ⁿ — 各回転可能結合のねじれ角
MATCHAはこれらをそれぞれ適切なリーマン多様体(Riemannian manifold)4上で定式化し、フローマッチング(flow matching)3によって各多様体の幾何学的制約を明示的に守りながらリガンドを「流し込む」設計です。多様体の構造に沿って動くため、生成されたポーズが物理的に破綻しにくいということらしいです。
2. 論文のメソッドとリポジトリ実装の概要
❑ MATCHAパイプライン全体像
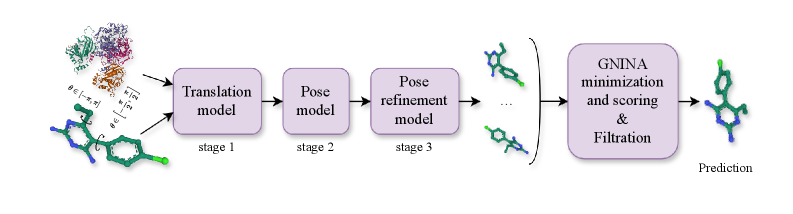
図1:MATCHAの3段階フローマッチングパイプライン。
Stage 1〜3を経てリガンドが結合ポーズへ「流れ込む」様子。
出典:MATCHA論文 (Frolova D. et al., 2025)
2.1 多段階精緻化(Multi-Stage Refinement):粗から密へ
MATCHAの中核戦略は、独立した3つのニューラルネットワークを順次適用する「粗から密へ(Coarse-to-Fine)」のアプローチです。
- Stage 1(Coarse):ランダムな位置から出発し、リガンドが結合ポケット付近へ大まかに移動します。平行移動の精度を最優先に学習したモデルが担当します。
- Stage 2(Medium):Stage 1の出力を初期値として受け取り、平行移動の精度をさらに高めながら、回転(SO(3))のサンプリングを開始します。
- Stage 3(Fine): Stage 2 の出力を受け取り、全自由度(平行移動・回転・ねじれ角)を最終的に微調整します。
2.2 リーマン・オイラー法による積分:euler() 関数の解剖
フローマッチングの推論とは、学習済みモデルが予測した「速度ベクトル場」を数値積分し、ノイズだらけの初期状態から正しいポーズへリガンドを「流す」作業です。utils/inference.py の euler() 関数がその積分を担当しています。3種の自由度それぞれで、異なる幾何学的更新則が使われています。
# utils/inference.py: euler() 関数(抜粋)
def euler(model, batch, device, num_steps=20):
cur_batch = deepcopy(batch).to(device)
h = 1. / num_steps # オイラー法のステップ幅(t=0→1を均等分割)
R_agg = cur_batch.ligand.init_rot # 初期回転行列
tr = cur_batch.ligand.init_tr # 初期平行移動ベクトル
tor = cur_batch.ligand.init_tor # 初期ねじれ角
for step in range(num_steps):
with torch.no_grad():
# モデルが各自由度の速度ベクトルを予測
dtr, drot, dtor, _ = model.forward_step(cur_batch)
# 【平行移動】ユークリッド空間: 単純な加算
tr = tr + h * dtr
# 【回転】SO(3): 指数写像で接空間→回転群に変換してから適用
R = expm_SO3(drot, h) # Lie代数 → 回転行列
# 【ねじれ角】SO(2)ⁿ: 周期性を考慮した更新
tor_agg += h * dtor
# 全自由度をバッチに一括適用
apply_tor_changes_to_batch_inplace(cur_batch, tor, is_reverse_order=False)
apply_tr_rot_changes_to_batch_inplace(cur_batch, tr, R)
cur_batch.ligand.t += h # 時刻 t を進める(0 → 1)
return cur_batch, tr, R_agg, tor_agg, pos_hist, trajectory
utils/rotation.pyのexpm_SO3(drot, h) 関数によって、リガンドの結合距離や角度を固定したまま、純粋な回転だけをサンプリングできます。
2.3 速度予測モデル:DiTと空間アテンションバイアスの融合
各ステージで速度ベクトルを予測するモデルは、画像生成で実績のある Diffusion Transformer (DiT) をベースに、分子ドッキング固有の工夫を2つ加えた構造になっています。
① 2つの CLSトークン:リガンドの原子埋め込みの先頭に2つの特殊トークン、CLS-0 :平行移動の速度予測用、CLS-1:回転の速度予測用が挿入されています。
② 空間的アテンションバイアス:Uni-Mol に着想を得た get_distance_bias() メソッドが、原子間の3次元距離をアテンション行列に直接注入します。このバイアス項がその物理的制約をモデルに学習させています。
タンパク質の入力特徴量には ESM-2-35M から抽出したアミノ酸残基埋め込みが使用されています。
3. MATCHAの使用方法と物理的妥当性の担保
MATCHAはシングルコマンドで動作する CLI(matcha)を提供しており、インストールも比較的シンプルです。以下の手順でパイプライン全体を試すことができます。
# シングルリガンドのドッキング実行例
matcha \
-r receptor.pdb \ # タンパク質構造(PDB形式)
-l ligand.sdf \ # リガンド構造(SDF形式)
-o results/ \ # 出力ディレクトリ
--n-samples 20 # 生成するポーズ数(デフォルト40)
GNINA によるエネルギー最小化とCNN親和性スコアリングと、PoseBusters5 の物理的妥当性フィルタが適用され、「原子が遠すぎる」「内部衝突がある」などのポーズが自動的に除外されます。
❑ 性能比較グラフ
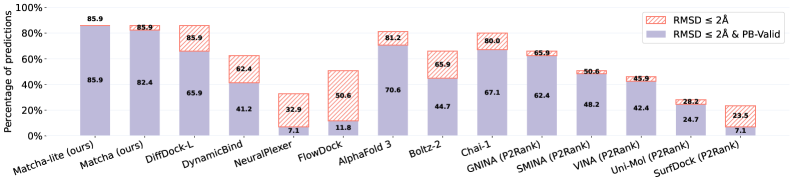
図3:DiffDock、AlphaFold3、Chai-1等とのブラインド・リガンドドッキングの性能比較。
出典:MATCHA論文 (Frolova D. et al., 2025)
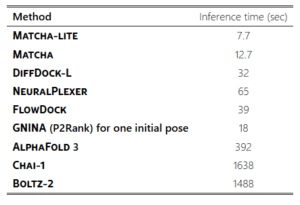 表1:DiffDock、AlphaFold3、Chai-1等とのブラインド・ドッキングモデルの平均推論時間の比較。
表1:DiffDock、AlphaFold3、Chai-1等とのブラインド・ドッキングモデルの平均推論時間の比較。
出典:MATCHA論文 (Frolova D. et al., 2025)
論文では、MATCHAはAlphaFold3やChai-1に匹敵する物理的妥当性(PoseBusters通過率)を達成しながら、推論速度は約31倍高速という結果が報告されています。創薬などでのインシリコスクリーニングへの応用が期待されます。
4. まとめと将来展望
MATCHAは「リーマン多様体上のフローマッチング」という数学的基盤と、「粗から密への多段階精緻化」という工学的設計を組み合わせることで、分子ドッキングの三大課題(速度・精度・物理的妥当性)をまとめて解決しようとするパイプラインです。
今後の展望として、タンパク質の柔軟性(induced fit)の考慮、RNA やペプチドといった他の生体分子系への応用などが挙げられます。
参考文献
- Corso G. et al. (2023) DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking. ICLR 2023. arXiv:2210.01776
- Frolova D. et al. (2025) MATCHA: Multi-stage Riemannian Flow Matching for Accurate and Physically Valid Molecular Docking. GitHub: https://github.com/LigandPro/Matcha
- Lipman Y. et al. (2022) Flow Matching for Generative Modeling. ICLR 2023. arXiv:2210.02747
- Chen Z. et al. (2023) Riemannian Flow Matching on General Geometries. ICML 2024. arXiv:2302.03660
- Buttenschoen M. et al. (2024) PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. Chem. Sci. doi: 10.1039/D3SC04185A

コメント