
このカテゴリーの記事では、「Pythonを使った機械学習やケモ・バイオインフォマティクスの実装や論文」を紹介しています。
本記事では、前回紹介したSMILESを事前学習したモデル「ChemBERTa」を使用して、ファインチューニングによる溶解度予測モデルを作ってみました。さらに、同じデータを用いて、DeBERTaベースの事前学習済みモデルのファインチューニングと、DeepChemのGCNによる予測も行ってみましたので紹介します。
ChemBERTaについては以下の記事に書いておりますので、読んでいただければ嬉しいです。

はじめに
前回紹介したChemBERTaでは、RoBERTa の実装に基づいて化合物の構造情報(SMILES)を用いて事前学習を行っています。この事前学習済みモデルは、Hugging Faceで公開されており、ラベル付きデータでファインチューニングすることが可能です。
今回は、ラベル付きデータとして化合物の溶解度データを用いて、その予測モデルを作成し、精度を検証してみたいと思います。
・事前学習モデルについて
Simple Transformersを用いることで、Hugging Faceで公開されている事前学習済み言語モデルを手軽に使用することができます。今回使用する事前学習済みモデルは、7700万個の化合物のSMILESの情報を事前学習させたものです(※1)。こちらは、ChemBERTa-2として論文がarXivに公開されています。
※1 Hugging FaceのChemBERTaモデルhttps://huggingface.co/DeepChem/ChemBERTa-77M-MLM
・ラベル付き(溶解度)データについて
GitHubにあるcurated-solubility-datasetのデータを使用させていただきました(※2)。ID, Name(化合物名), SMILES, Solbilityなど 9983個のデータが含まれています。
※2 溶解度データ保存Github https://github.com/whitead/dmol-book/tree/main/data
ChemBERTaのファインチューニング
以下https://dmol.pub/dl/pretraining.htmlを参考にさせて頂き、Google colabolatoryでファインチューニングを実行しました。
必要なパッケージやライブラリーを準備します。
#事前学習モデルを手軽に使用できます。
!pip install simpletransformers#ライブラリのインポート
from simpletransformers.classification import ClassificationModel
import pandas as pd, sklearn, matplotlib.pyplot as plt, numpy as np
from sklearn.model_selection import train_test_split
from sklearn.metrics import mean_absolute_error, mean_squared_errorラベル付きデータを予測モデル用に整えます。
# データセットを読み込む
soldata = pd.read_csv(
"https://github.com/whitead/dmol-book/raw/main/data/curated-solubility-dataset.csv"
)
# データを学習用とテスト用に分割(train/test=8/2)
train, test = train_test_split(soldata, test_size=0.2, shuffle=True, random_state = 42)
# 学習とテストデータセットを整形
train_dataset = train[["SMILES", "Solubility"]] #SMILESとSolubilityの情報を抽出
train_dataset = train_dataset.rename(columns={"Solubility": "labels", "SMILES": "text"})
test_dataset = test[["SMILES", "Solubility"]]
test_dataset = test_dataset.rename(columns={"Solubility": "labels", "SMILES": "text"})
test_dataset | text | labels | |
| 1253 | CCc1ccc2C(=O)c3ccccc3C(=O)c2c1 | -5.98424 |
| 9930 | CC1OC(OC2C(O)CC(OC3C(O)CC(OC4CCC5(C)C(CCC6C5CC… | -4.16 |
| 7046 | OC(=O)c1ccccc1c2ccccc2C(O)=O | -2.2839 |
| 8711 | ClC(=C(C=C(Cl)Cl)Cl)Cl | -4.23 |
| 6880 | CO[P](=O)(OC)OC | 0.5526 |
| … | … | … |
| 2249 | [Na+].[Na+].[Na+].[Na+].[Na+].COc1cc(N=Nc2cc(c… | -2.48339 |
| 210 | C/C=C/C(=O)C1C(C)C=CCC1(C)C | -3.39637 |
| 4961 | CCc1ccccc1Br | -3.6702 |
| 1919 | [K+].OC(=O)c1ccccc1C([O-])=O | -0.39102 |
| 718 | Cc1ccc(N)cc1Cl | -1.75313 |
使用する事前学習済みモデルとその学習方法を指定し、実際に学習させます。
# 回帰モデルを設定、DeepChem/ChemBERTa-77M-MLMを使用
model = ClassificationModel(
"roberta",
"DeepChem/ChemBERTa-77M-MLM",
num_labels=1,
args={
"num_train_epochs": 30,
"regression": True,
"use_multiprocessing_for_evaluation": True,
"train_batch_size": 16,
"save_eval_checkpoints": False,
"save_model_every_epoch": False,
"output_dir": "outputs_chemBERTa/"
},
use_cuda=True,
)#モデル学習
model.train_model(train_df=train_dataset)# モデル評価、結果の表示(テストデータを用いたモデルの評価)
result, model_outputs, wrong_predictions = model.eval_model(test_dataset)
print(result){‘eval_loss’: 1.2386267702877523}
# 学習データに対する予測
predictions_train = model.predict(train_dataset["text"].tolist())[0]
# 実際の溶解度(Actual Solubility)と予測値(Predicted Solubility)をプロットし、相関係数を計算
plt.figure(figsize=[6,6])
plt.scatter(train_dataset["labels"].tolist(), predictions_train, color="C0")
plt.plot(train_dataset["labels"], train_dataset["labels"], color="C1")
plt.text(-8,-12,
f"Correlation coefficient: {np.corrcoef(train_dataset['labels'], predictions_train)[0,1]:.3f}",
fontsize = 14)
plt.xlim(-13.5, 3)
plt.ylim(-13.5, 3)
plt.xlabel("Actual Solubility")
plt.ylabel("Predicted Solubility")
plt.show()
# 平均絶対誤差 (MAE)
mae = mean_absolute_error(train_dataset['labels'], predictions_train)
print("MAE for Train:", '{:.3f}'.format(mae))
# 平均二乗誤差 (MSE)
mse = mean_squared_error(train_dataset['labels'], predictions_train)
print("MSE for Train:", '{:.3f}'.format(mse))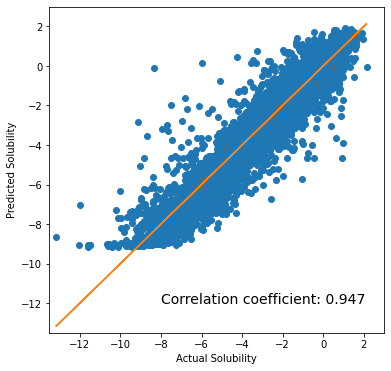
MAE for Train: 0.589 MSE for Train: 0.661
# テストデータに対する予測
predictions = model.predict(test_dataset["text"].tolist())[0]
# 実際の溶解度Xと予測値Yをプロットし、相関係数を計算
plt.figure(figsize=[6,6])
plt.scatter(test_dataset["labels"].tolist(), predictions, color="C0")
plt.plot(test_dataset["labels"], test_dataset["labels"], color="C1")
plt.text(-8,-12,
f"Correlation coefficient: {np.corrcoef(test_dataset['labels'], predictions)[0,1]:.3f}",
fontsize = 14)
plt.xlim(-13.5, 3)
plt.ylim(-13.5, 3)
plt.xlabel("Actual Solubility")
plt.ylabel("Predicted Solubility")
plt.show()
# 平均絶対誤差 (MAE)
mae = mean_absolute_error(test_dataset['labels'], predictions)
print("MAE:", '{:.3f}'.format(mae))
# 平均二乗誤差 (MSE)
mse = mean_squared_error(test_dataset['labels'], predictions)
print("MSE:", '{:.3f}'.format(mse))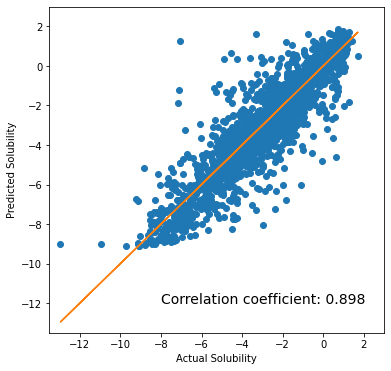
MAE: 0.783 MSE: 1.240
まとめ
| Correlation efficient | MAE | MSE | |
| Train | 0.947 | 0.589 | 0.661 |
| Test | 0.898 | 0.783 | 1.24 |
上記のように、SMILESから溶解度を予測可能なモデルを簡単に構築することができました。精度はそこそこという感じでしょうか。学習データに対する精度に対して、テストデータに対する精度が低く、今回の学習データや学習方法では過学習となっています。「化学」や「分子」を表現する空間は非常に広大と考えられ、今回の学習データ量でも十分とは言えない可能性もありますし、 SMILESを事前学習していることの限界なのかもしれません。
DeBERTaベースのモデルとGCNによる予測
次に、同じ溶解度データについて、異なる2つの手法で予測モデルを作ってみます。一つは、DeBERTaを基にした事前学習モデルで、上記と同様の手順でファインチューニングを行い、予測モデルを作成しました。もう一つは、グラフ畳み込みニューラルネットワーク(GCN)についてラベル付きデータで学習し予測モデルを作成してみました。
◆DeBERTa
DeBERTaは、RoBERTaと同じく自然言語処理のモデルです。固定されたマスキング方法が使用されているRoBERTaとは異なり、DeBERTaでは動的なマスキング方法が採用されており、多様なトークン表現を学習できるといわれています。また、RoBERTaでは、テキスト中の位置情報が考慮されていませんが、DeBERTaでは位置情報を考慮した Disentangled attentionメカニズム が採用されています。このように、RoBERTaの改良版という感じのモデルです。
DeBERTa論文:DeBERTa: Decoding-enhanced BERT with Disentangled Attention https://arxiv.org/abs/2006.03654
色々と勉強している際に、DeBERTaベースのSMILESを事前学習したモデルとして、sagawa/PubChem-10m-deberta が公開されていることに気づき、使用させていただきました。以下のHuggin Faceにて公開されています。また、Qiitaにその詳細が解説されています。
参考サイト 事前学習モデル:https://huggingface.co/sagawa
解説:https://qiita.com/luddite/items/3323696ddae1236bf918
ChemBERTaと同様に、溶解度データを学習とテストに分け、事前学習済みモデルをファインチューニングし、予測モデルを評価しました。
◆グラフ畳み込みニューラルネットワーク(GCN)
GCNは、グラフ構造に機械学習の手法を適用するグラフニューラルネットワーク(GNN Scarselliらによって2009年に報告)の1つです。分子の化学構造はグラフとして表現可能ですので、分子の性質の予測に応用されています。
GCN論文:Convolutional Networks on Graphs for Learning Molecular Fingerprints https://arxiv.org/abs/1509.09292
GCNを行うツールとして、以前紹介したDeepChemを使用しました。特徴量として ConvMolFeaturizer を使用することで、SMILESをGCNの入力とし、GraphConvModelにてモデルを作成しました。上記と同様に、溶解度データを学習データとテストデータに分けて、学習を行い、モデルを評価しました。
以下にDeepChemで溶解度データを扱った記事をあげています。こちらもみていただけると嬉しいです。
以下に、3つの手法で得られた溶解度予測モデルの精度をまとめました。
| Correlation efficient | MAE | MSE | ||
| RoBERTa | Train | 0.947 | 0.589 | 0.661 |
| Test | 0.898 | 0.783 | 1.24 | |
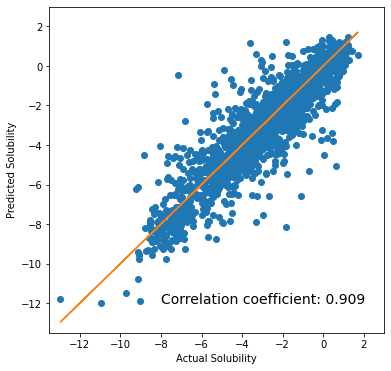
| DeBERTa | Train | 0.996 | 0.238 | 0.11 |
| Test | 0.909 | 0.686 | 1.045 | |
| GCN | Train | 0.943 | 0.665 | 0.769 |
| Test | 0.897 | 0.848 | 1.311 |
結果として、RoBERTaをベースとしたChemBERTa-2に比べて、DeBERTaをベースとしたsagawaさんの sagawa/PubChem-10m-deberta がより良い精度を示しました。一方で、DeBERTaベースのモデルでも過学習の傾向があり、やはり学習に用いた溶解度データが足りないのでしょうか。それともSMILESの限界でしょうか。
また、事前学習済みモデルを使っていないことを考えれば、GCNも健闘といえるのではないでしょうか。
SMILESではなくグラフ構造を入力とし、グラフ構造の事前学習ができると、さらなる精度の向上が期待できるのではと考えられます。この辺り、グラフのTransformerの研究も進められています。


コメント