
このカテゴリーの記事では、「Pythonを使った機械学習やケモ・バイオインフォマティクスの実装や論文」を紹介しています。
今回の記事では、自然言語処理モデルBERTを分子へと応用した、ChemBERTaを紹介したいと思います。
はじめに
これまで、タンパク質のドッキングや分子動力学シミュレーション、ケモインフォマティクス的な手法をざっくばらんに紹介してきました。近年、ニューラルネットワークはさまざまに応用されています。例えば、以下で紹介した“EquiBind” では、 タンパク質の結合位置とリガンド(小分子)の結合ポーズの探索に、深層学習が適用されています。

深層学習(Deep learning)は多種多様なモデル・ネットワークが開発されています。例えば、画像判別におけるConvolutional Neural Networks (CNN) や自然言語処理におけるAttention、Transformer等です。特に、Transformerを基礎にもつモデルは爆発的に発展しています。2022年に公開され非常に話題になっている、チャットAI ChatGPT もその1つです。また、グーグルDeepMind社のタンパク質の立体構造予測AlphaFoldも、素晴らしい性能を発揮しています。これらのモデルは、様々なタスクに特化しているわけですが、高い性能を発揮できる共通の理由として、これらが大規模言語モデル(LLM: Large Language Model)であるという点があります。
LLMにおいては、大規模な文章データが用意され、そのテキストに出てくる単語(トークン)の関係性が学習されます。この学習は自己教師的な事前学習として実施されています。どのように自己教師を設定するかですが、よく使われているのは、用意した文章データに登場する単語の何割かをマスクして隠し、その隠された単語を予測する方法 (masked language model) です。このような事前学習済みモデルは、大規模に与えられた文章データ同士や、登場する単語同士の持つ関係性・意味合いをおおよそ把握した状態になっていると考えて良さそうです。また、用いる文章や単語に対して、あらかじめ人間が何等かの意味合いや数値を設定しているわけではなく、文章や単語の関係性や意味合いの理解について、人間を超える可能性がある気もします(素人ですので、誤解していたらすみません(๑´ڡ`๑))。また、文章データを事前学習させればさせるほど、高い性能を発揮することができるため、いわゆるビックデータが必要となります。
LLMをタンパク質に応用することを考えれば、「大規模な文章データ」は「多くのタンパク質のアミノ酸配列(一次構造)」ということになり、「単語(トークン)」は「アミノ酸」ということになります。 立体構造予測AlphaFoldだけでなく、Meta社のタンパク質言語モデル Evolutionary Scale Modeling(ESM)などが公開され、高い性能を示しています。タンパク質の場合、近年の次世代シーケンス(NGS)技術の発展にともなって、様々な生物のゲノム情報が明らかとなっており、そのタンパク質の一次構造情報が蓄積されている、即ち、ビックデータを用意できるようになっているわけですね。スゴイ時代です!
今回紹介する “ChemBERTa” は、化学におけるLLMです。その実装が公開され、以下の論文がarXivにあがっています。また、さらに大規模モデルとなったChemBERTa-2も公開されています。
●ChemBERTa 論文:ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction https://arxiv.org/abs/2010.09885
●ChemBERTa-2 論文: ChemBERTa-2: Towards Chemical Foundation Models https://arxiv.org/abs/2209.01712
ChemBERTa論文の紹介
本記事では、ChemBERTa 論文に記載されています、ChemBERTaの概要とその性能とこれからの可能性について触れながら紹介してたいと思います。
本論文では、自然言語処理タスクにおいて高い性能を発揮するTransformerを分子物性予測タスクに適用した、ChemBERTa が実装されています。ChemBERTaにおいて、分子のSMILES表記が自然言語の文章に相当します。
方法
ChemBERTaは、RoBERTa の実装に基づいています。RoBERTaは、自然言語処理モデルとして広く使用されているBERTの発展版です。BERTは、先述のmasked language modelに加え、next sentence prediction(次の文章の予測)などをタスクとした事前学習を行うことにより、精度が向上したモデルです。RoBERTaは、BERTと比べ、事前学習データセット数と回数を増やしたり、より長い文章を学習させたりすることで、精度が向上したモデルであるといわれています。基本的なアーキテクチャ(計算の骨組み)の詳細は以下(1)をご参照ください。今回は、RoBERTaのTransformerの設定は、12個のattention heads(self-attention (どこに注目すべきかの推測 下記の図のMulti-Head attentionはself-attentionを複数用意したもの))×6層で72個のattentionメカニズムから構成されています。
アーキテクチャに関する参考文献(1) 下記画像引用
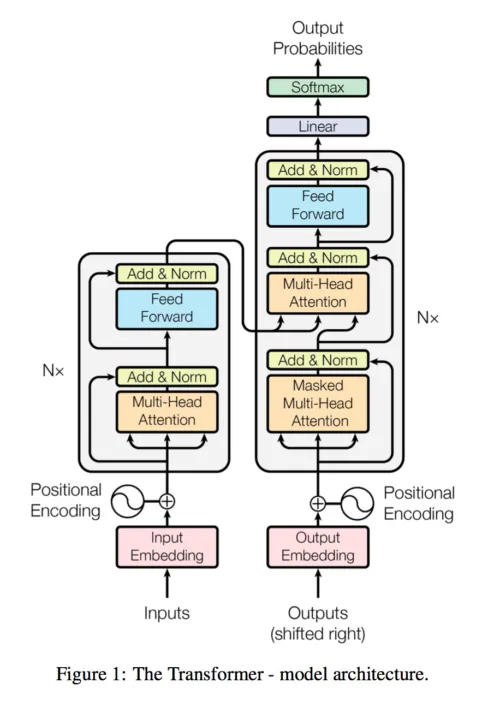
Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N Gomez, Łukasz Kaiser, and Illia Polosukhin. 2017. Attention is all you need. In Advances in neural information processing systems. https://arxiv.org/pdf/1706.03762.pdf
著者らは、PubChemの7700万分子のSMILESデータを用いて、単語(トークン、SMILESで定義される元素記号および結合や電荷等を示す記号)の関係性を事前学習させました。
さらに、事前学習したモデルを用いて、別のラベル付きデータで個別のタスクにファインチューニングし、その性能を評価しています。ラベル付きデータは、MoleculeNet※から、SMILESの化学構造に対する、BBBP(血液脳関門の通過性(透過性)を表す2値ラベルデータ)やClin Tox(臨床毒性(CT_TOX)), HIV(HIVの複製を阻害する能力), Tox21(p53ストレス応答経路活性化(SR-p53))を使用しました。
① Directed Message Passing Neural Network (D-MPNN GNNの新たなモデル) 、ランダムフォレスト(RF)、サポートベクタマシン(SVM)など他の手法での予測精度との比較や、②事前学習データのデータサイズによる予測精度への影響、③Byte-Pair Encoding (BPE)やカスタムSmilesTokenizerなどの異なるトークンナイザーとの比較、④SMILESとSELFIES表現の性能差の評価、⑤Attentionの可視化、などの結果が報告されています。
※MoleculeNet:分子特性などを含む機械学習方法をテストするために特別に設計されたデータセットです。70万以上の化合物の特性に関するデータを含んでいます。DeepChemの開発者グループによって開発されたものであり、DeepChemから使うこともできます。
結果
①ChemBERTaと他の手法での予測精度との比較
各ラベル付きデータを用いたD-MPNN、RF、SVMの手法での予測モデルに対し、ChemBERTaでの予測モデルは、予測精度を大幅に改善することはできませんでした (Table1)。
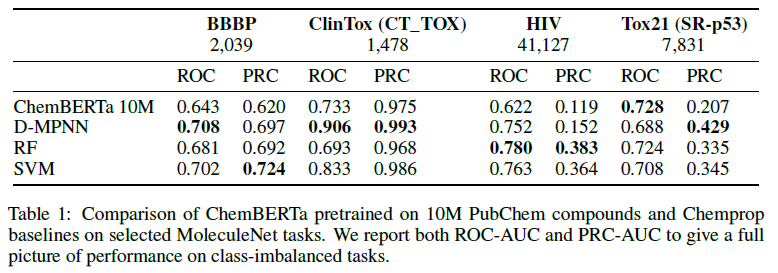
ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction より引用
②事前学習データのデータサイズによる予測精度への影響
事前学習データサイズの違いによって、その後のファインチューニングによって得られるモデルの予測精度が変化しました(Fig.1)。ChemBERTaの性能は、事前学習データサイズに比例して向上しています。
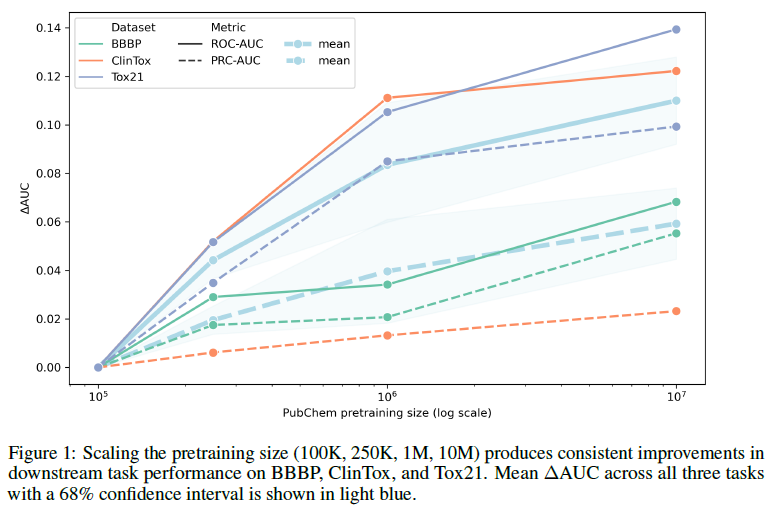
ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction より引用
③トークナイザーの比較
HuggingFace tokenizers libraryのByte-Pair Encoder (BPE)に対し、ChemBERTaで使用するSmilesTokenizerを用いた結果を比較しました。PubChem-1Mセットで2つの同じモデルを事前学習し、Tox21 SR-p53タスクで精度を評価しました。その結果、SmilesTokenizerはPRC-AUC = +0:015でBPEをわずかに上回っていました。
④SMILESとSELFIES表現の性能差
SMILESとSELFIES(機械学習用に設計された代替分子文字列表現)のそれぞれで事前学習を行いました。事前学習の結果を用いて、Tox21 SR-p53タスクで精度を評価しましたが、統計学的有意差は確認できませんでした。
⑤ Attentionの可視化
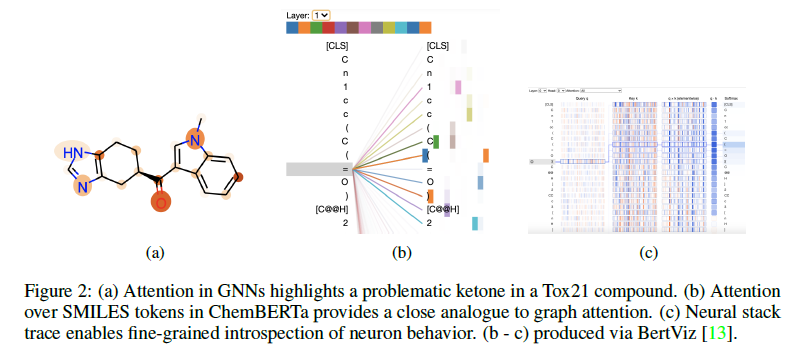
BertViz(TransformerのAttention可視化ツール) を使用して、Tox21上のChemBERTa(SmilesTokenizerバージョン)のAttention headを可視化し、 グラフニューラルネットワーク(GNN)の分子グラフのAttentionの可視化と対比しています(Fig.2 例としてケトン基に着目)。化学的に強く関連する官能基や芳香環を確認でき、AttentionベースのRNNの既報とも一致することが分かりました。
ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction より引用
考察
ChemBERTaは、ほとんどのタスクで最先端技術を上回る性能を発揮できていませんが、将来的には、事前学習のスケールアップ、より大きなマスキング率の実験、マルチタスクのファインチューニング、ELECTRAのような代替事前学習手法の研究などでの向上が期待される、とのことです。
ここまで見て来たように、ChemBERTaでは、SMILESのビッグデータを事前学習データとして用いて、そのトークン間の関係性をTransformerで学習しています。この手法は、自然言語処理やタンパク質に対しては、大きな成功を収めていますが、ChemBERTaでは、そこまでのインパクトはなかったようです。1つの大きな問題は、そもそもSMILES表記によって、各分子の特性をどれほど表現できているのかという点が挙げられると思います。自然言語では単語(トークン)の並び方、タンパク質ではアミノ酸の並び方、「配列そのもの」が、文章やタンパク質の実体そのものをかなりの程度表現することになります。それに対して、SMILESという記号の並び方では、分子の実体を表現する力が足りないということかもしれません。
現在、より分子の表現に適切であると思われる 分子グラフとTransformerの利点を組み合わせたハイブリッドモデルなどについても研究が進んでいるとのことで(Graphormer や こちらなど)、さらなる性能向上が期待されています。


コメント