
このカテゴリーの記事では、「Pythonを使った機械学習やケモ・バイオインフォマティクスの実装や論文」を紹介しています。Python は3系(3.7)、anacondaを中心にして環境構築しています。
● CPU Intel® Core™ i7-6700K CPU @ 4.00GHz × 8
● メモリ 64GB
● GPU GeForce GTX 1080/PCIe/SSE2
● OS CentOS Linux 8 stream or Ubuntu 20.04.1 LTS
● Python 3.7
今回の記事では、深層学習によるタンパク質と小分子の結合予測を実行することができるEquiBindをインストールし、実行しました。CentOS 8、Ubuntu 20.04の両OSで同様の手順で実施できることを確認しています。
はじめに
タンパク質とタンパク質、タンパク質と小分子(リガンド)がどのように結合するのか、実証したり予測したりすることは、基礎研究および創薬等の応用研究にとって重要な課題です。予測の手法として、いわゆるドッキングシミュレーション(本ブログでも“LightDock” 、“AutoDock Vina”を紹介しています。)が広く用いられてきましたが、近年、深層学習を応用し、結合予測を行う試みが進んでいます。以下の記事では、“AutoDock Vina”およびそのフォークである“SMINA”のスコアリング関数を深層学習により向上させた、“GNINA”のインストールとテストの実行を紹介しました。

GNINAでは、結合ポーズのスコアリングに深層学習を適用していますが、タンパク質のどこにリガンドが結合するのか(結合位置)、リガンドがどのような向きで結合するのか、これらの配座空間の探索については、伝統的な手法(遺伝的アルゴリズムやモンテカルロ法)となっています。詳細は原著論文をご参照ください。
●GNINA論文:A McNutt, P Francoeur, R Aggarwal, T Masuda, R Meli, M Ragoza, J Sunseri, DR Koes. (2021) GNINA 1.0: molecular docking with deep learning J Cheminform. 2021; 13: 43. doi: 10.1186/s13321-021-00522-2
さらなる発展として、タンパク質の結合位置とリガンドの結合ポーズ、これらの探索自体に深層学習を適用することで、より高い精度で、より高速に予測を実施できる可能性があり、期待されています。ディープラーニング凄い!
本記事では、そのような深層学習的手法の1つとして、“EquiBind” を紹介します。こちらは、以下の論文がarXivに公開されていますが、2022年5月の時点では査読中のようです。
●EquiBind 論文: Hannes Stärk, Octavian-Eugen Ganea, Lagnajit Pattanaik, Regina Barzilay, Tommi Jaakkola EquiBind : Geometric Deep Learning for Drug Binding Structure Prediction https://arxiv.org/abs/2202.05146
深層学習(Deep learning)は画像判別、囲碁や将棋などのゲーム、自然言語処理、タンパク質立体構造予測(AlphaFoldなど)等の様々な分野で素晴らしい成果を上げていますが、その研究過程で、多種多様なモデル・ネットワークが開発されています。例えば、画像判別におけるConvolutional Neural Networks (CNN) や自然言語処理におけるAttention、Transformer等です。また、これらモデル技法は、他の課題に流用することができ、AlphaFoldではTransformerの技法が使われています。今回紹介する “EquiBind” では、Graph Matching Networks (GMN) と E(3)-equivariant Graph Neural Networks (E(3)-GNN) が用いられているとのことですが、その詳細は原著論文を参照してください(๑´ڡ`๑)
本記事では、EquiBindをインストールした後、GNINAの記事で実行したテストランと同様のタンパク質-リガンド複合体について、ドッキング予測を行ってみました。
EquiBind のインストール
GitHubに記載されている手順通り、condaでインストールを行いました。下記を順に実行してください。
git clone https://github.com/HannesStark/EquiBind cd EquiBind conda env create -f environment.yml
一行目でGitリポジトリをクローンすることで、ファイルをダウンロードしています。次に出来たEquiBindディレクトリに移動し、environment.ymlを引数としてconda env create を実行します。
これで仮想環境 equibind ができて、インストールも行われています。とても簡単です。
EquiBind によるドッキング予測の実行
それでは早速、EquiBind を使ってみましょう。まずは下記を実行し、仮想環境equibindをアクティブにしましょう。
conda activate equibind
テストランとして、GNINAの記事でも取り上げた、MAP KINASE ERK2(タンパク質)とSB220025(リガンド)の複合体構造(PDBID:3ERK)を用いたドッキング予測をやってみましょう。詳細は、記事やPDBのリンク等を参照してください。
EquiBindディレクトリ下にdataディレクトリがありますが、その中にドッキングに使用するファイルを準備するようにしたいと思います。下記を順に実行します。
cd data #EquiBindディレクトリの中にあるdataディレクトリに移動 mkdir inputs #inputsディレクトリを作成 cd inputs mkdir 3ERK cd 3ERK wget http://files.rcsb.org/download/3ERK.pdb #PDBより3ERK.pdbファイルをダウンロード grep ATOM 3ERK.pdb > 3ERK_protein.pdb #3ERK.pdbファイルのタンパク質部分を3ERK_protein.pdbとして保存 grep SB4 3ERK.pdb > 3ERK_ligNoH.pdb #3ERK.pdbファイルのリガンド部分を3ERK_ligNoH.pdbとして保存
EquiBind/dataディレクトリ下にinputs/3ERKディレクトリを用意し、その中に3ERK.pdbファイルを取得しました。grepコマンドを使い、3ERK.pdbファイルのタンパク質部分、リガンド部分をそれぞれpdbファイルとして保存しました。
ここで、EquiBindでは、ドッキング予測に用いるリガンド分子は、水素原子を含む完全な構造情報を有している必要があります(タンパク質の方は水素原子を含まなくても大丈夫です。)。多くのPDBファイルには、水素原子の座標情報が含まれていませんので、リガンドの適切な位置に水素原子を付加してやる必要があります。ここでは obabel を用いて水素原子を付加しました。このコマンド(openbabel)は、EquiBindが依存するプログラムとして、仮想環境equibind に同時にインストールされているはずです。下記を実行してください。
obabel 3ERK_ligNoH.pdb -O 3ERK_ligand.pdb -h #3ERK_ligNoH.pdbに水素を付加して3ERK_ligand.pdbとして保存
3ERK_ligand.pdb を確認してもらえば、水素原子が付加されていることがわかると思います。obabel の使い方の詳細はリンク等を参照してください。これで、タンパク質とリガンドのファイルが用意できましたが、これらのファイル名は、hoge_protein.pdbとhoge_ligand.pdbとする必要があり、さらにこれらをhogeディレクトリに入れておく必要があります(今回は、hogeを3ERKとしています。)。
最後に、EquiBindディレクトリ下のconfigs_cleanディレクトリにあるinference.ymlを書き換え、ファイルの場所(3ERKディレクトリがある場所)を指定してやる必要があります。inference.ymlを開き、下記のようにinference_path :に’data/inputs’を記載して上書き保存してください。

これでドッキングを実行できます。EquiBindディレクトリまで戻り、下記を実行してください。
python inference.py --config=configs_clean/inference.yml
うまくいけば、以下のような出力が得られると思います。
[2022-05-08 17:12:19.305814] [ Using Seed : 1 ] Processing 3ERK: complex 1 of 1 Trying to load data/inputs/3ERK/3ERK_ligand.pdb Docking the receptor data/inputs/3ERK/3ERK_protein.pdb To the ligand data/inputs/3ERK/3ERK_ligand.pdb Writing prediction to data/results/output/3ERK/lig_equibind_corrected.sdf Saving predictions to runs/flexible_self_docking/predictions_RDKitFalse.pt
ドッキング予測で得られたリガンドの結合ポーズは、dataディレクトリ下にresults/output/3ERK/lig_equibind_corrected.sdf として保存されます。lig_equibind_corrected.sdfをPyMOLで読み込み、結果を表示してみましょう。PyMOLインストールは以下の記事を参考にしてください。
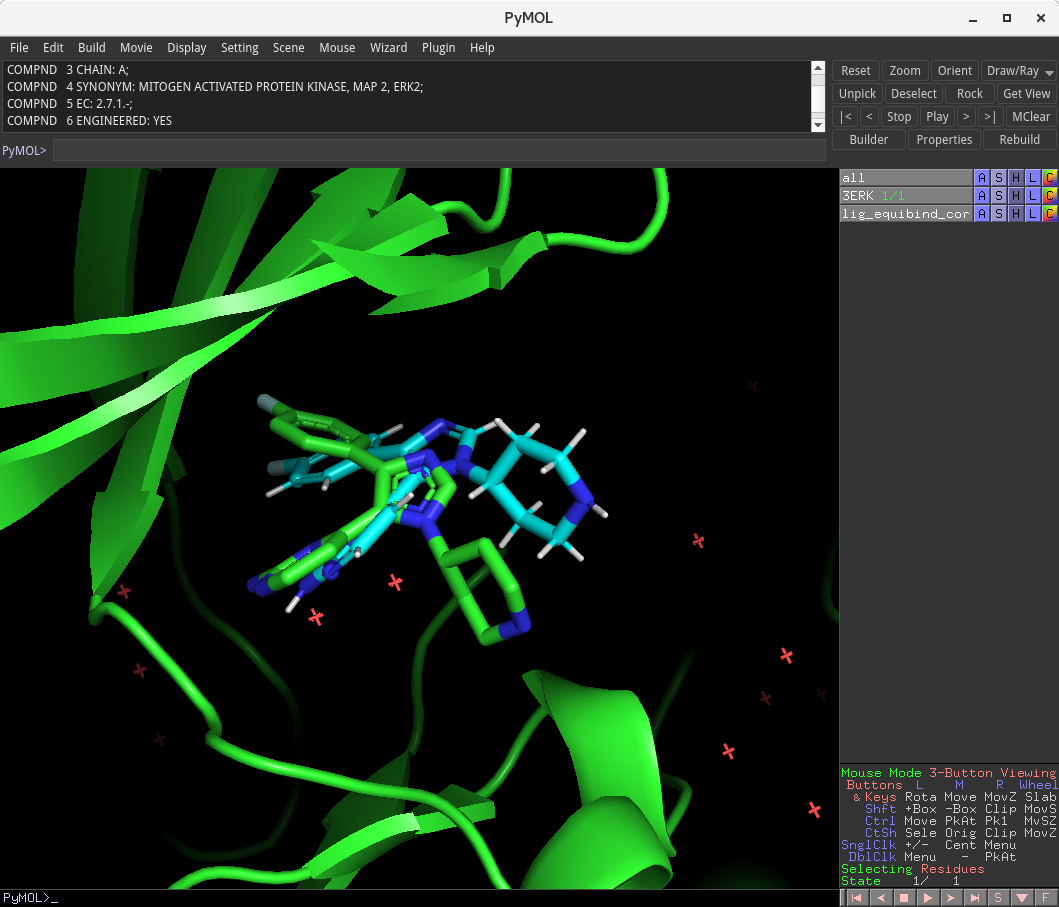
以下を実行して、複合体構造 3ERK.pdbと今回の予測結果を読み込みます。
pymol data/inputs/3ERK/3ERK.pdb data/results/output/3ERK/lig_equibind_corrected.sdf #EquiBindディレクトリで実行します
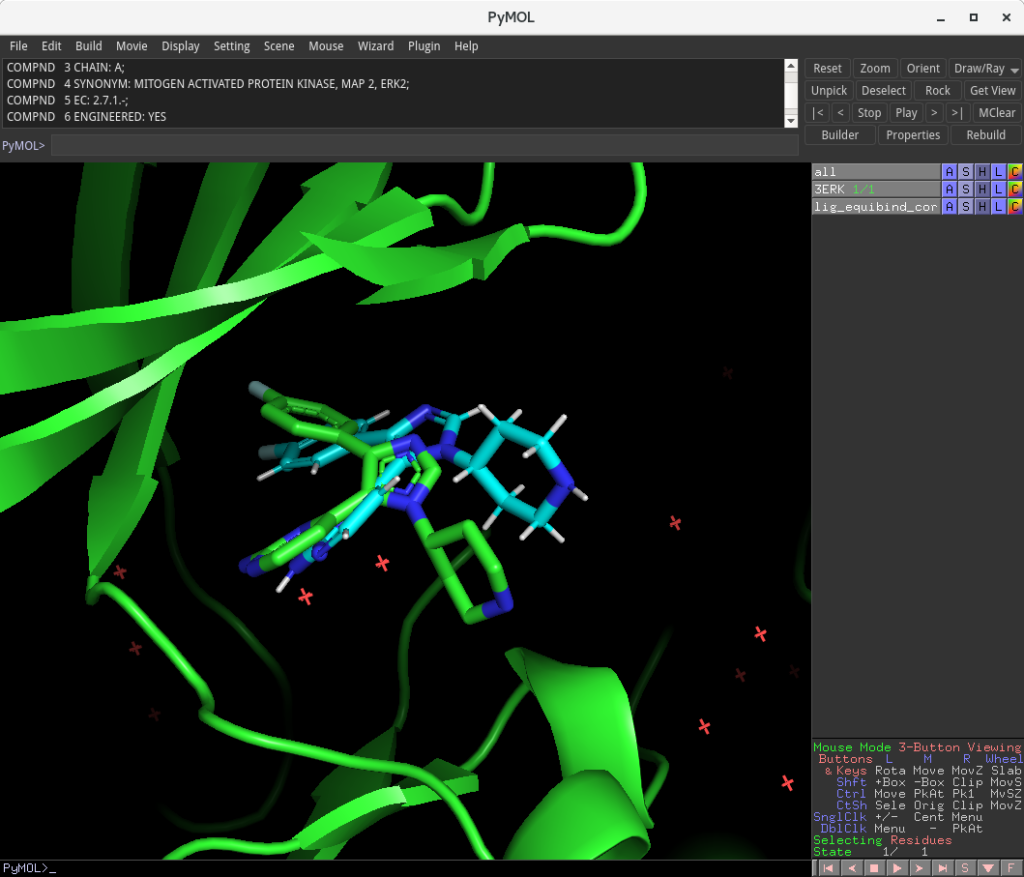
上のような表示が得られたかと思います。緑色が複合体構造(PDBID:3ERK)、水色がEquiBindで予測された結合ポーズです。GNINAの実行例と比べると、複合体構造のポーズと少しズレていますが、タンパク質の結合位置、リガンドの配向はおおよそ合致していると言ってよいでしょう。深層学習によって、結合位置やドッキングポーズを概ね再現できているようです。ディープでポン!(๑´ڡ`๑)
ただし今回用いた複合体構造3ERKは、実はEquiBindの学習データとしても使用されています。深層学習では、過学習(過剰適合)の問題がありますので、未知の複合体予測に使えるか検証が必要な気がします。
EquiBindのインストールと結合予測を試すことができました。一般に深層学習による予測は高速に実行可能ですので(学習には時間がかかりますが)、多数のタンパク質-リガンドペアを網羅的にドッキングすることも可能だと考えられます。さらなる発展に期待ですね!

コメント