
このカテゴリーの記事では、「Pythonを使った機械学習やケモ・バイオインフォマティクスの実装や論文」を紹介しています。Python は3系(3.7)、anacondaを中心にして環境構築しています。
● CPU Intel® Core™ i7-6700K CPU @ 4.00GHz × 8
● メモリ 64GB
● GPU GeForce GTX 1080/PCIe/SSE2
● OS CentOS Linux 8
● Python 3.7
今回の記事では、タンパク質 – 小分子のドッキングシミュレーションソフトウェアGNINAのインストールを行いました。その際、CentOSでは必要なライブラリを認識させることが難しく、公式ページ通り、Ubuntu にインストールしました。anacondaのインストールや使い方は同じです。他の記事も参考にしてください。
● OS Ubuntu 20.04.1 LTS
● Python 3.8
はじめに
これまでに、タンパク質-タンパク質間、タンパク質-ペプチド間、タンパク質-DNA間のドッキングシミュレーションを実施するためのソフトウェアとして“LightDock” を紹介しました。開発者から記事にコメント頂き、嬉しかったです(・(ェ)・)
また、タンパク質-小分子のドッキング用途として“AutoDock Vina”(2021年にバージョンアップ)を紹介しました。
タンパク質のドッキングシミュレーションは、酵素の基質認識、創薬等、基礎研究ならびに応用研究に用いられてきましたが、近年、他のバイオインフォマティクス手法と同様、機械学習(深層学習)でその精度を向上させようという試みが進んでいます。本記事では、“AutoDock Vina”およびそのフォークである“SMINA”のスコアリング関数を深層学習により向上させた、“GNINA”のインストールを行ってみたいと思います。それぞれ以下の論文で報告されている手法です。スコアリング関数は、リンクのwikipediaで大変詳しく解説されているように思いますので、参考にしてください(๑´ڡ`๑)
●AutoDock Vina 論文: O. Trott and A. J. Olson. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry, 31(2), 455-461.
●SMINA 論文: Koes DR, Baumgartner MP, Camacho CJ. (2013) Lessons learned in empirical scoring with smina from the csar 2011 benchmarking exercise. J Chem Inform Model. 53(8):1893–1904.
●GNINA論文:A McNutt, P Francoeur, R Aggarwal, T Masuda, R Meli, M Ragoza, J Sunseri, DR Koes. (2021) GNINA 1.0: molecular docking with deep learning J Cheminform. 2021; 13: 43. doi: 10.1186/s13321-021-00522-2
GNINAは、GitHubでソースコードやインストール方法が公開されています。インストールするためには、cmakeによるコンパイルが必要ですが、記載されている方法に沿って実行すれば大丈夫です。
GNINA のインストール
インストール手順は、GitHubの通りで問題ないと思いますが、今回、Ubuntuシステム上のPythonに変更を加えたくなかったため、下記のように仮想環境 gninaを用意し、Pythonに関連するパッケージはcondaでインストールしました。また、cmakeもcondaで仮想環境にインストールしました。condaは色々な環境を用意できるので、便利ですね。
sudo apt install libboost-all-dev libatlas-base-dev libhdf5-dev librdkit-dev #様々なライブラリをインストール sudo apt install git wget libgoogle-glog-dev libprotobuf-dev protobuf-compiler #様々なライブラリをインストール conda create -n gnina python=3.8 #python 3.8 の仮想環境 gninaを作成 conda activate gnina #仮想環境 gninaをアクティブに conda install pytest numpy cmake #pytest, numpy, cmakeのインストール conda install python-devtools -c conda-forge #python-devtools のインストール
そのうえで、以下のように、gitでOpenbabel3のソースコードをダウンロードし、インストールします。buildディレクトリをつくり、そのなかでcmake, make, make installと順番に実行します。
git clone https://github.com/openbabel/openbabel.git cd openbabel git checkout openbabel-3-1-1 mkdir build cd build cmake -DWITH_MAEPARSER=OFF -DWITH_COORDGEN=OFF -DPYTHON_BINDINGS=ON -DRUN_SWIG=ON .. make sudo make install
これにより、OpenBabel 3.1.0がインストールされました。ここで、端末を終了して、新たに立ち上げてください。そうすると、obabelコマンドが実行できるようになっていると思います。
再度、仮想環境gninaをアクティブにして、以下のように、GNINAをインストールしましょう。Openbabel3と同じようにcmake, make, make installを実行します。
conda activate gnina #仮想環境 gninaをアクティブに git clone https://github.com/gnina/gnina.git cd gnina mkdir build cd build cmake .. make sudo make install
GNINAのコンパイルには少し時間がかかるかもしれませんが、以上でインストール完了です。端末を終了して、新たに起動すれば、gninaコマンドが使用できるようになっているはずです。
GNINA によるドッキングの実行
それでは早速、GNINAを使ってみましょう。公式の実行例がcolab notebookに作成されています。この例では、MAP KINASE ERK2(タンパク質)とSB220025(小分子)の複合体構造(PDBID:3ERK)のPDBファイルからタンパク質と小分子を抜き出し、それらのドッキングを行います。この複合体構造の詳細については、PDBのリンク等を参照してください。複合体状態の構造からタンパク質と小分子を別々に抜き出し、それらが元の位置に戻るかどうかを試すシミュレーションとなっています。登録された構造に戻れば、正解ということになります。
リンクを参考に、以下を順に実行します。
mkdir gnina_test #テストランを実行するディレクトリgnina_testを作成 cd gnina_test wget http://files.rcsb.org/download/3ERK.pdb #PDBより3ERK.pdbファイルをダウンロード grep ATOM 3ERK.pdb > rec.pdb grep SB4 3ERK.pdb > lig.pdb
grepコマンドを使うことで、タンパク質をrec.pdb、小分子をlig.pdbで保存しています。これでドッキングの準備ができました。
gninaコマンドを以下のように実行することで、ドッキングシミュレーションが開始されます。
gnina -r rec.pdb -l lig.pdb --autobox_ligand lig.pdb -o docked.sdf --seed 0
_
(_)
__ _ _ __ _ _ __ __ _
/ _` | '_ \| | '_ \ / _` |
| (_| | | | | | | | | (_| |
\__, |_| |_|_|_| |_|\__,_|
__/ |
|___/
gnina master:8943ed1 Built May 6 2022.
gnina is based on smina and AutoDock Vina.
Please cite appropriately.
Commandline: gnina -r rec.pdb -l lig.pdb --autobox_ligand lig.pdb -o docked.sdf --seed 0
==============================
*** Open Babel Warning in PerceiveBondOrders
Failed to kekulize aromatic bonds in OBMol::PerceiveBondOrders
Using random seed: 0
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
***************************************************
mode | affinity | CNN | CNN
| (kcal/mol) | pose score | affinity
-----+------------+------------+----------
1 -8.52 0.9024 6.788
2 -8.09 0.6081 6.603
3 -8.31 0.4515 6.454
4 -6.62 0.3029 6.010
5 -6.24 0.2846 6.096
6 -6.83 0.2695 5.776
7 -6.86 0.1569 5.462
8 -6.76 0.1438 5.844
9 -6.16 0.1354 5.330
上のようなアウトプットが表示され、数秒でシミュレーションが終了しました。*** Open Babel Warning in PerceiveBondOrders との出力がありますが、とりあえず良しとします(๑´ڡ`๑) CNN pose scoreの良い順に1〜9のポーズが出力されています。このスコア関数が深層学習(CNN)の適用で改良されており、良いシミュレーションポーズが得られるようになっているのでしょう。
結果について、PyMOLで表示してみましょう。インストールは以下の記事を参考にしてください。
以下のpymolコマンドを実行して、複合体構造 3ERK.pdbと今回のシミュレーション結果を読み込みます。
pymol 3ERK.pdb docked.sdf
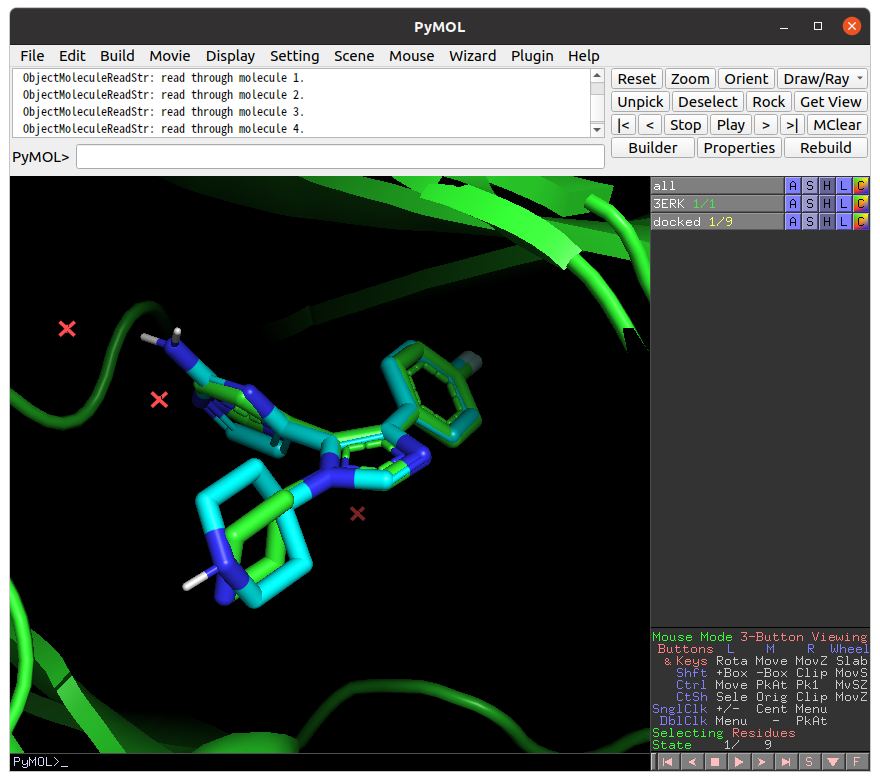
上のような表示が得られたかと思います。緑色が複合体構造(PDBID:3ERK)、水色がシミュレーションのトップスコアの構造です。ほとんど一致していると言ってよいでしょう。
GNINAのインストールとテストシミュレーションを実行し、正解の複合体構造を再現できました。AutoDock Vinaによるシミュレーションも実行し、両者の結果を比較してみると面白いかもしれませんね。今回はここまでとしたいと思います。

コメント