
このカテゴリーの記事では、「Pythonを使った機械学習やケモ・バイオインフォマティクスの実装や論文」を紹介していきたいと考えています。Python は3系(3.7)、anacondaを中心にして環境構築していきます。以下のようなハード・ソフト環境(CentOS Linux)を用いますが、Python(anaconda)が利用できれば、異なる環境下でも類似の実装が可能かと思います。
- CPU Intel® Core™ i7-6700K CPU @ 4.00GHz × 8
- メモリ 64GB
- GPU GeForce GTX 1080/PCIe/SSE2
- OS CentOS Linux 8
- Python 3.7
LightDock の実行 ーComplex protein-protein docking with restraintsー
前回の記事で 仮想環境 LightDockを設定し、インストールも済んでいることを前提として進めます。まずは、conda activate LightDock を実行して、環境 LightDockをアクティブにしてください。
1. 受容体とリガンドの立体構造データのダウンロード
その上で、ホームディレクトリに test ディレクトリを作成し、その中にチュートリアル用のデータをダウンロードしましょう。次のコマンドを順番に実行します。
$ mkdir test $ cd test $ curl -O https://raw.githubusercontent.com/lightdock/lightdock.github.io/master/tutorials/examples/4G6M/4G6M_rec.pdb $ curl -O https://raw.githubusercontent.com/lightdock/lightdock.github.io/master/tutorials/examples/4G6M/4G6M_lig.pdb
このチュートリアルでは、2つのタンパク質分子同士のドッキングシミュレーションを実行します。それらは、ヒトのインターロイキン1β というタンパク質と、その抗体である gevokizumab です。上記のcurl コマンドによって、 gevokizumab が 4G6M_rec.pdbとして、インターロイキン1β が4G6M_lig.pdb としてダウンロードされます。gevokizumab は抗体の全長ではなく、インターロイキン1βへの結合領域のみの構造が利用されます。タンパク質ータンパク質間のドッキングシミュレーションでは、より大きな方を「受容体(receptor)」、小さな方を「リガンド(ligand)」として設定することが多く、今回のチュートリアルでは、gevokizumab の構造( 4G6M_rec.pdb )を「受容体」、インターロイキン1β の構造(4G6M_lig.pdb)を「リガンド」として扱っていきます。
2. 残基拘束データの作成(ダウンロード)
LightDockでは、ドッキングシミュレーションを様々な設定で実行可能です。例えば、様々な生化学的実験や立体構造の情報から、受容体およびリガンドがどのような領域やアミノ酸残基で相互作用するか推定できた場合、その推定した情報を「残基拘束(residue restraints)」として使用することが可能です。このチュートリアルでは、以下のコマンドで restraints.list をダウンロードして、残基拘束を用いたシミュレーションを実行します。test ディレクトリ で コマンドを実行してください。
$ curl -O https://raw.githubusercontent.com/lightdock/lightdock.github.io/master/tutorials/examples/4G6M/restraints.list
ダウンロードされた restraints.list はテキストファイルで、下記のような情報が書き込まれています。
R A.GLN.27 R A.SER.30 R A.TYR.32 ・ ・ ・ L B.SER.68 L B.CYS.69 L B.VAL.70 ・ ・ ・
最初の ”R” は受容体を示しており、gevokizumab のうち、 27番GLN(グルタミン)、 30番SER(セリン)、 32番TYR(チロシン)・・・が相互作用するアミノ酸残基として指定されます。途中から”L”で始まる行が始まりますが、ここからはリガントとして扱うインターロイキン1β のアミノ酸残基を指定しています(68番SER(セリン)、 69番CYS(システイン)、 70番VAL(バリン)・・・が相互作用する。)。この記述様式に従ってアミノ酸残基を指定することで、様々な残基拘束条件を与えることが可能です。
3. ドッキングシミュレーション条件の設定
上記で、test ディレクトリに3つのファイル(受容体4G6M_rec.pdb, リガント4G6M_lig.pdb, 残基拘束restraints.list)がダウンロードできました。これらを指定する形で、下記のコマンドを実行します。
$ lightdock3_setup.py 4G6M_rec.pdb 4G6M_lig.pdb 400 200 -anm --noxt --noh -rst restraints.list
これにより、400個の初期 スワーム(swarms) と、そのswarmごとに200個のグローワーム(glowworms)を生成するドッキングシミュレーションのための設定が実行されます。この設定実行が正常に終了すると、複数のファイルとディレクトリが作成されます。また、400 個 swarms を作る設定を実行しましたが、残基拘束条件を与えているため、実際に生成された swarms は84個となりました。*swarms と glowworms の詳細は論文や開発者のサイトを参照して下さい。
これら swarms が受容体(gevokizumab)のどの領域に設定されたかを確認してみましょう。コマンド実行でinit ディレクトリが作成されていると思います。以前の記事で PyMOLをインストールしていれば、別途ターミナルを立ち上げて、下記のコマンドを実行してみてください。
$ conda activate pymol $ pymol lightdock_4G6M_rec.pdb ./init/cluster_centers.pdb
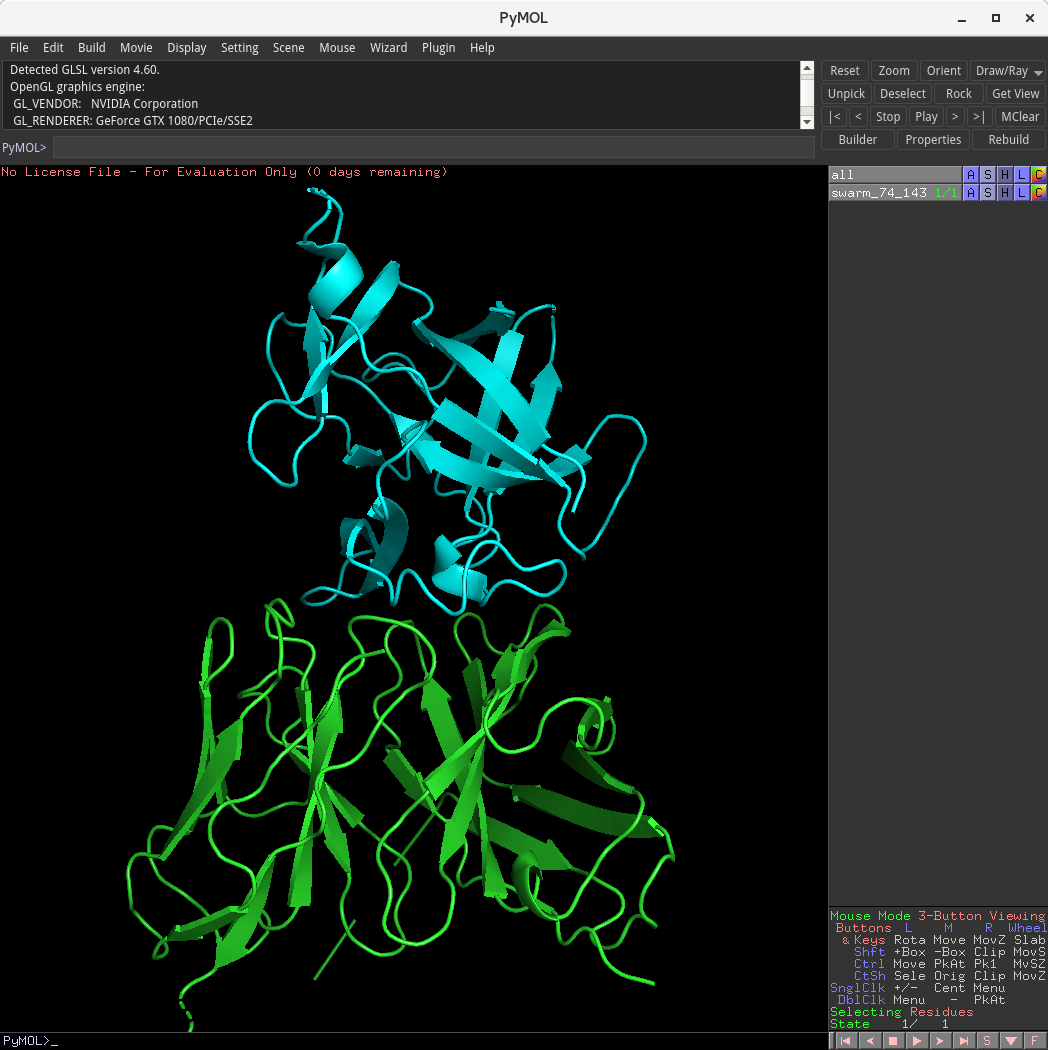
1行目で、仮想環境 pymol をアクティブにしています。2行目でpymol を起動すると同時に、設定で作成された受容体 gevokizumab の構造ファイル lightdock_4G6M_rec.pdb とinit ディレクトリ内に作成された84個のswarmsの座標を示すファイル cluster_centers.pdb を読み込んでいます。
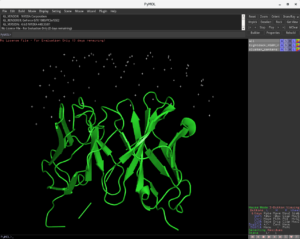
上図において、緑色のリボンモデルで受容体 gevokizumab が示されており、その上部に散在している白いマークが84個のswarmsの位置です。また、これら84カ所それぞれに、200個のglowworms が設定されており、次のステップで、それらの位置にリガンド(インターロイキン1β )がドッキングされることになります。
4. ドッキングシミュレーションの実行
次のコマンドを実行することで、上記設定のシミュレーションが始まります。
$ lightdock3.py setup.json 100 -s fastdfire -c 8
引数の 100 は GSO optimization を100ステップ行うということを指定しています。-s fastdfire で各ドッキングポーズ(端的に言えば、どれほど安定な相互作用様式が得られているか)を評価するスコアリング関数をDFIRE に指定しています。また、-c 8 で 8 コア並列の計算を実行するように指定していますので、パソコンのCPUコア数に応じて変更してください。
[lightdock] INFO: simulation parameters saved to ./lightdock.info
[lightdock_setup] INFO: Ignoring OXT atoms
[lightdock_setup] INFO: Ignoring Hydrogen atoms
[lightdock_setup] INFO: Reading structure from lightdock_4G6M_rec.pdb PDB file...
[lightdock_setup] INFO: 1782 atoms, 230 residues read.
[lightdock_setup] INFO: Ignoring OXT atoms
[lightdock_setup] INFO: Ignoring Hydrogen atoms
[lightdock_setup] INFO: Reading structure from lightdock_4G6M_lig.pdb PDB file...
[lightdock_setup] INFO: 1194 atoms, 149 residues read.
[lightdock] INFO: Loading scoring function...
[lightdock] INFO: Using DFIRE scoring function
[lightdock] INFO: Done.
[kraken] INFO: Kraken has 8 tentacles (cpu cores)
[kraken] INFO: Tentacle ready with 11 tasks
[kraken] INFO: Tentacle ready with 11 tasks
[kraken] INFO: Tentacle ready with 11 tasks
[kraken] INFO: Tentacle ready with 11 tasks
[kraken] INFO: Tentacle ready with 10 tasks
[kraken] INFO: Tentacle ready with 10 tasks
[kraken] INFO: Tentacle ready with 10 tasks
[kraken] INFO: Tentacle ready with 10 tasks
[kraken] INFO: 84 ships ready to be smashed
[lightdock] INFO: Monster spotted
[kraken] INFO: Release the Kraken!
[0] step 1
[1] step 1
[2] step 1
[3] step 1
[4] step 1
[5] step 1
[6] step 1
[7] step 1
・
・
・
このような表示が出て、84個のswarms分、シミュレーション計算が流れていきます。パソコンスペックによりますが、数十分程度で計算が終了します。
5. シミュレーション結果の分析
計算が終了したら、以下の手順で結果を解析します。
(1) swarmごとにシミュレーション構造を生成します(swarmごとに200個の glowworms を設定していますので、200個ずつドッキング構造が生成されます。)
(2) swarmごとにシミュレーション構造をクラスター化する(近しい構造同士をまとめる。)
(3) ランキングファイルの作成(スコアリング関数で計算されたドッキング構造の評価値をまとめる。)
(4) 残基拘束を満した割合によるフィルタリング(40%以上を推奨)
この手順をまとめたスクリプトファイルを用意して、実行します(analysis.sh を圧縮したものをanalysis リンクからダウンロードできるようにしています。)。以下のコマンドは、ここまでシミュレーションを進めてきた test ディレクトリで実行して下さい。
$ bash analysis.sh
解析が終了すると、新たにfiltered ディレクトリが作成され、40%フィルタリングを満たすシミュレーション構造が保存されます。さらに、各ドッキング構造のスコアリング関数による評価値(高い程良い)のランキングもrank_filtered.list として作成されます。rank_filtered.list はテキストファイルですので、そのまま開いてください。
swarm_74_143.pdb 58.352 0.976
swarm_59_6.pdb 54.798 0.976
swarm_54_23.pdb 54.592 0.976
・
・
・
上記のように、ドッキング構造ファイル名、スコア、残基拘束を満した割合、が並んでいます。シミュレーションの度にわずかに結果は変わってきますが、今回の場合、58.352が最高スコアで、その構造がswarm_74_143.pdb で、filtered ディレクトリに保存されています。さきほどと同様に、PyMOLで結果を開いてみましょう。
$ pymol ./filtered/swarm_74_143.pdb #ファイル名は結果にあわせて換えること。
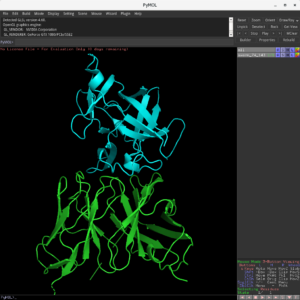
緑色のリボンモデルで受容体 gevokizumab が示されており、その上部に水色のリボンモデルでリガンド(インターロイキン1β )が結合した、ドッキングシミュレーション構造が得られています。
補足
gevokizumab とインターロイキン1β が結合した立体構造は、両分子が結合した状態での結晶構造解析が実施されており、PDB に ID:4G6M で登録されています。この構造が真の複合体構造と考えられます。PyMOLを使って4G6M の構造を表示・分析すれば、シミュレーション構造との差を比較することも可能ですが、今回の記事はこの辺で終わりたいと思います。
コメント
すごいチュートリアル、ありがとうございます
I’m very glad to hear from you. We have just started this blog for commercial and non-profit researchers in Japan. Please forgive us for linking to you without permission, and thank you again for your continuous support.
Very nice initiative, glad that LightDock software is useful for your research and customers. Of course, nothing to forgive as LightDock is open source and free software!
Thank you so much once again. ありがとうございます!