
このカテゴリーの記事では、「Pythonを使った機械学習やケモ・バイオインフォマティクスの実装や論文」を紹介していきたいと考えています。Python は3系(3.7)、anacondaを中心にして環境構築していきます。以下のようなハード・ソフト環境(CentOS Linux)を用いますが、Python(anaconda)が利用できれば、異なる環境下でも類似の実装が可能かと思います。
● CPU Intel® Core™ i7-6700K CPU @ 4.00GHz × 8
● メモリ 64GB
● GPU GeForce GTX 1080/PCIe/SSE2
● OS CentOS Linux 8
● Python 3.7
はじめに
今回、分子動力学法(Molecular dynamics)計算の実例として、Trp-cageを例に、タンパク質の折りたたみ(Protein Folding)シミュレーションを実行したいと思います。
Trip-cage の分子動力学シミュレーションの文献としては、下記などを参考にして下さい。
● J Am Chem Soc 2002 Sep 25;124(38):11258-9. doi: 10.1021/ja0273851. All-atom structure prediction and folding simulations of a stable protein Carlos Simmerling 1, Bentley Strockbine, Adrian E Roitberg
● PNAS November 11, 2003 100 (23) 13280-13285; https://doi.org/10.1073/pnas.2233312100 Trp-cage: Folding free energy landscape in explicit water Ruhong Zhou
● Chemistry Letters 2021, Vol.50, No.1 162-165 The Folding of Trp-cage is Regulated by Stochastic Flip of the Side Chain of Tryptophan Takunori Yasuda, Yasuteru Shigeta, and Ryuhei Harada https://doi.org/10.1246/cl.200699
シミュレーション系の設定や実際の演算は、OpenMM によって行います。OpenMM のインストールとテストについては、以下の記事を参考にしてください。

シミュレーションの初期構造、つまり折りたたむ前の(アンフォールド状態の)Trp-cageの分子構造については、以下の記事で構築していますので、参考にしてください。
OpenMM-Setup のインストール
OpenMMに付属するアプリケーションである OpenMM-Setup を用いることで、シミュレーションの設定を実施することができます。まずはこれをインストールしましょう。OpenMM をインストールしてある仮想環境 openmmをアクティブにしましょう。
$ cd OpenMM #OpenMMディレクトリに移動 $ conda activate openmm #仮想環境openmm をアクティブに
この環境下に conda でインストールします。こちらのサイトの内容も参照してください。実際には、以下のコマンドを実行するだけです。
$ conda install -c conda-forge openmm-setup
これでインストール完了です。続いて設定に移ります。
OpenMM-Setup によるシミュレーションの設定
それではOpenMM-Setup によってシミュレーションの設定を行いましょう。以下を実行してください。
$ openmm-setup
下のように、OpenMM-Setup のGUI がウェブブラウザ上で起動します。
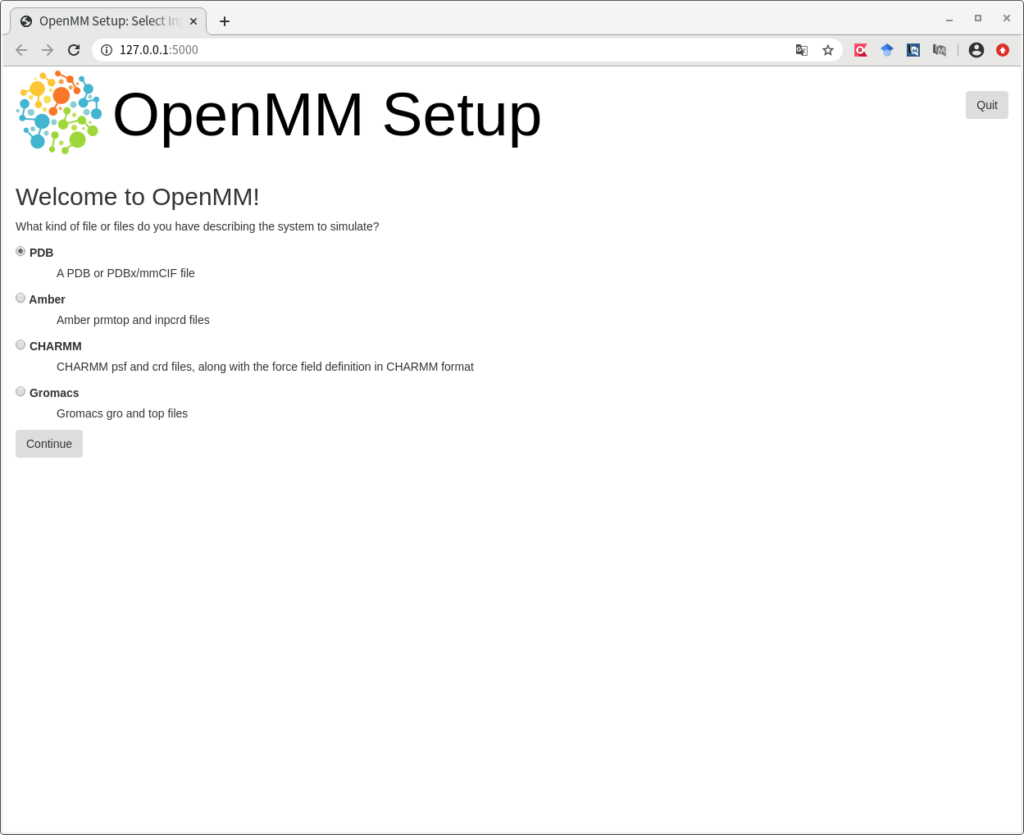
このブラウザ上で、設定を進めていきます。前回の記事で、折りたたむ前の(アンフォールド状態の)Trp-cageの分子構造をTrpCage.pdb で保存しています。これをもとに設定を進めますので、上のようにPDB を選択したまま、Continue をクリックして進めます。
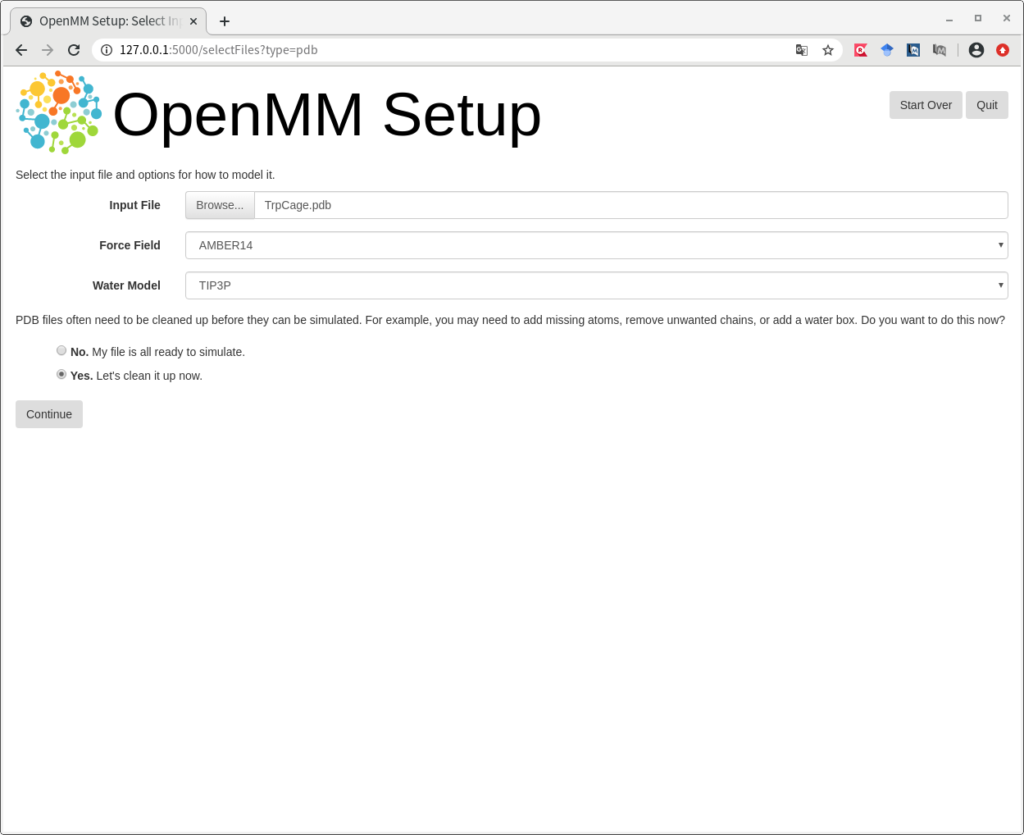
上のような表示になりますので、Input File のBrowse…から TrpCage.pdb を選択してください。また、Force Field は AMBER14 (初期設定のまま)を、Water Model は TIP3P を選択しました。Force Field や Water Model の詳細については、http://amber.tkanai-lab.org/ や https://ambermd.org/tutorials/ 等、Amber チュートリアルやマニュアルを参照してください。最後に、構造に問題がある場合に修正が行われるように、Yes. Let’s clean it up now. を選択し、Continue をクリックして進めてください。
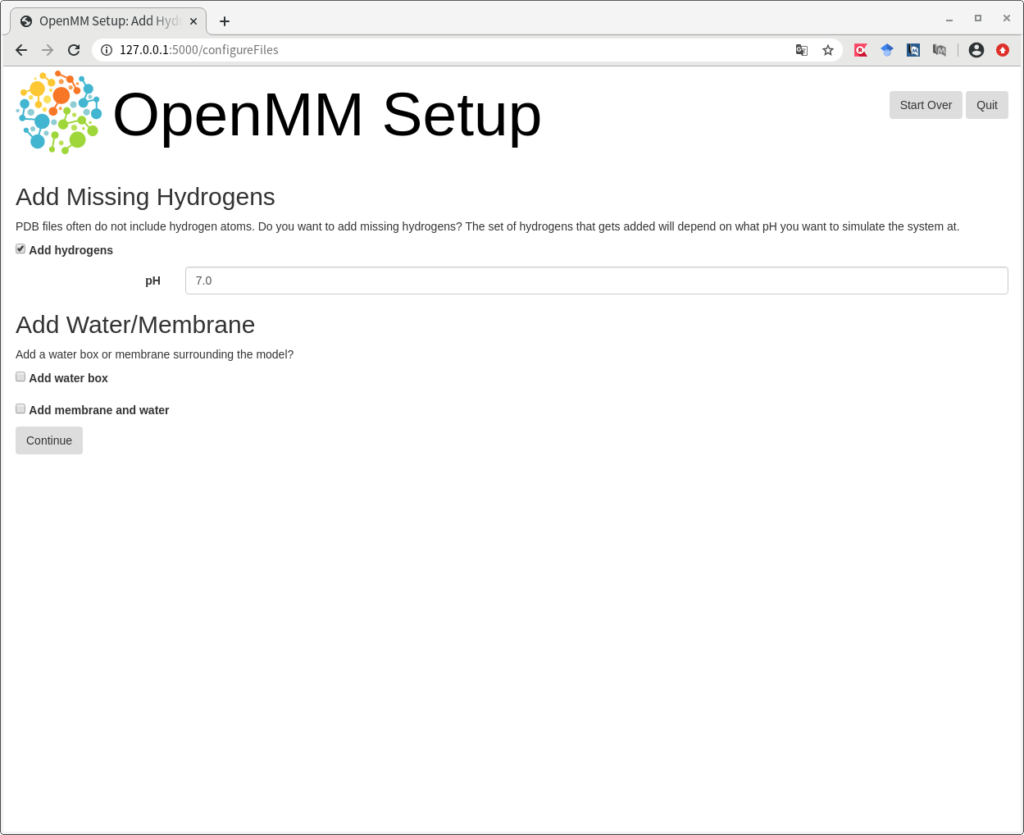
上の表示に切り替わります。上部の Add Missing Hydrogens はそのままの設定にしたいと思います。これで、Trp-cage 全20アミノ酸の正しい位置に、水素原子が付加されます。また、Add Water/Membrane は以下の画面のように設定してください。
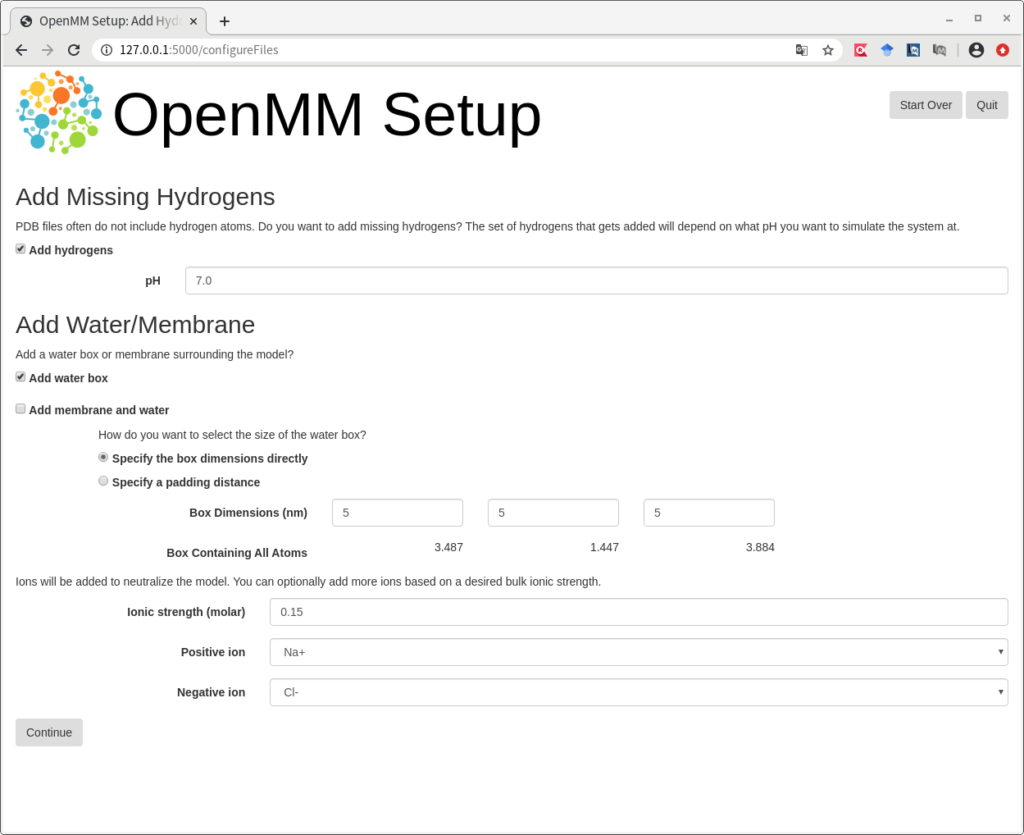
Add water box の方をチェックし、さらに、Specify the box dimensions directlyを選択し、Box Dimensions (nm) に 5 を入力し、5 x 5 x 5 nm3 立方体のシミュレーション box (周期境界条件)を設定しました。シミュレーション boxのサイズは、タンパク質の大きさによって変更する必要があります。今回、Trp-cage が20 アミノ酸残基と小さいため、この程度のサイズにしました。最後に、Ionic strength (molar) に 0.15 を入力し、NaCl を150 mM になるように配置する設定としました。これらの詳細についても、OpenMMのサイトに加えて、http://amber.tkanai-lab.org/ や https://ambermd.org/tutorials/ 等も参照してください。 Continue をクリックして進めます。
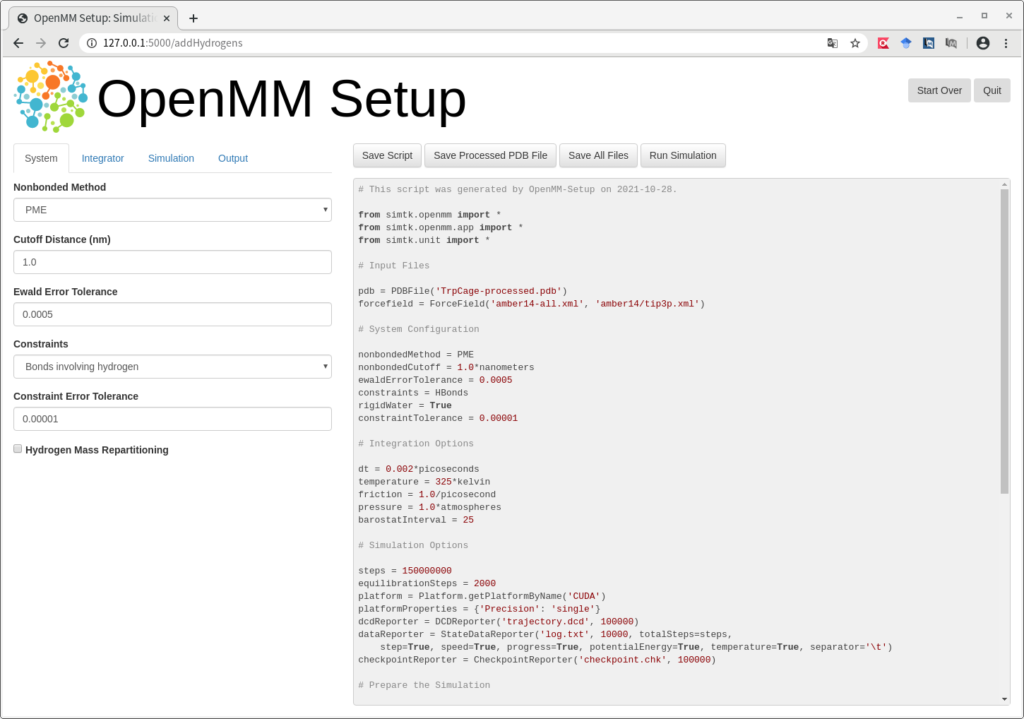
上の表示に切り替わります。上記のSystem、Integrator、Simulation、Output それぞれの設定を入力します。System は上記を参考にしてください。Integrator は以下のようにしました。
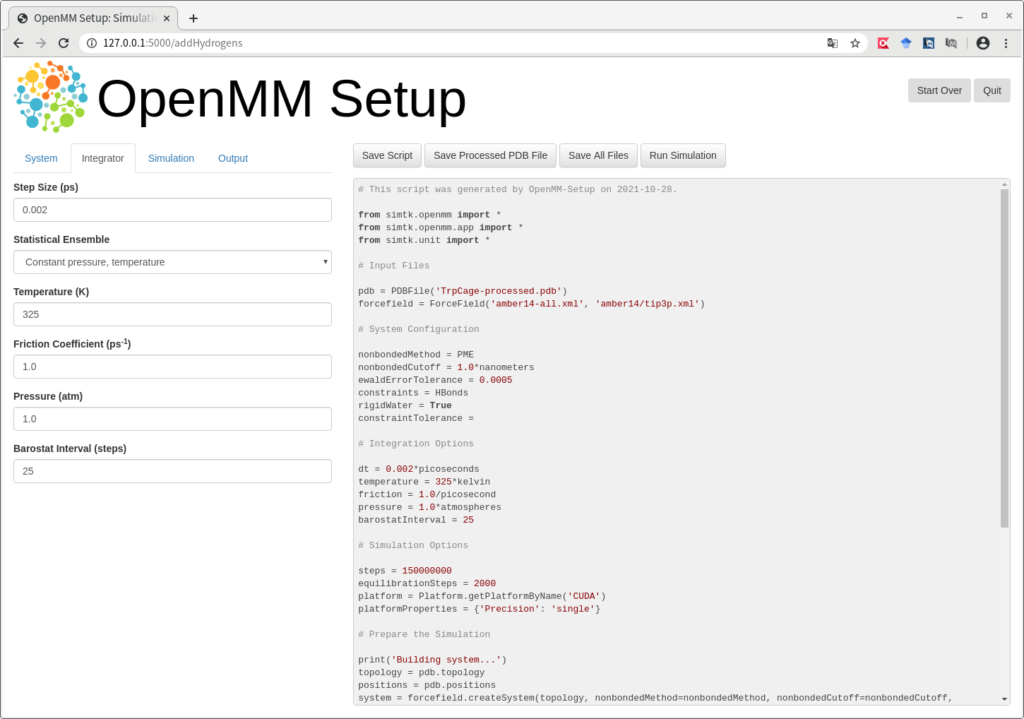
Step Size (ps)の初期値が0.004 となっていますが、こちらは0.002 ps に変更しました。また、Temperature (K)は325K (52℃に相当)と高めにしています。これは、http://ambermd.org/tutorials/basic/tutorial3/ のAmber チュートリアルを参考にしていますので、リンクを確認ください。Simulation は以下のように設定しました。
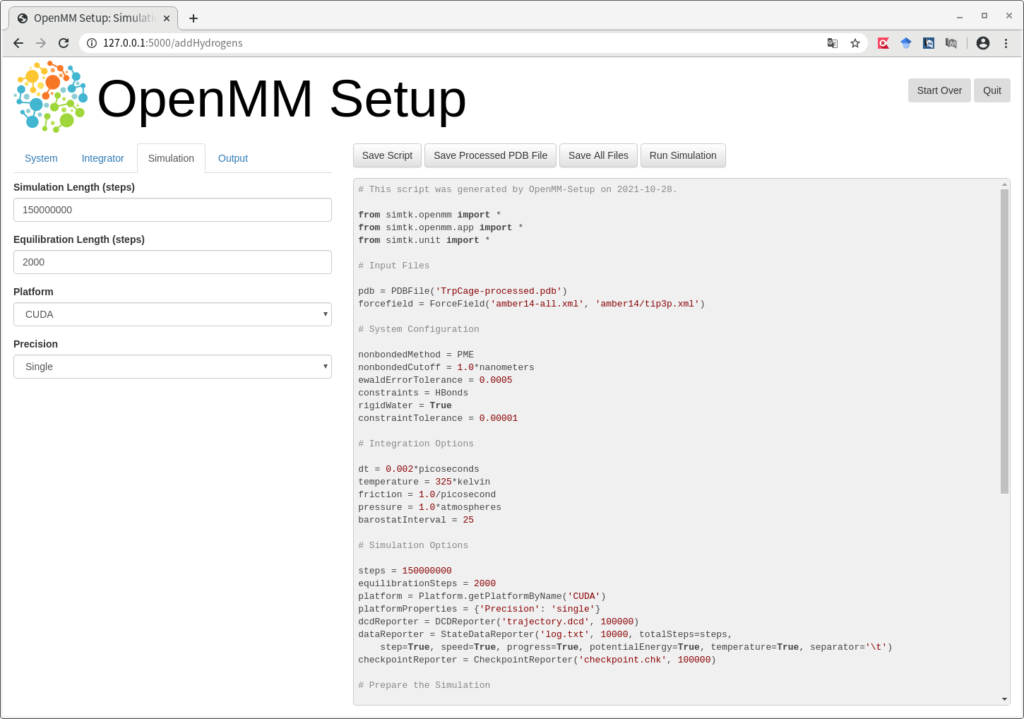
Simulation Length (steps) は150000000 としました。この設定で、0.002 ps × 150000000 = 300000 ps = 300 ナノ秒 間のシミュレーションが可能です。また、Platform は CUDA となっており、これで GPU を用いた計算が可能です(*GPU として GeForce GTX 1080 を使用した場合、計算終了まで約20時間が必要でしたので、実行時は注意してください。) 。
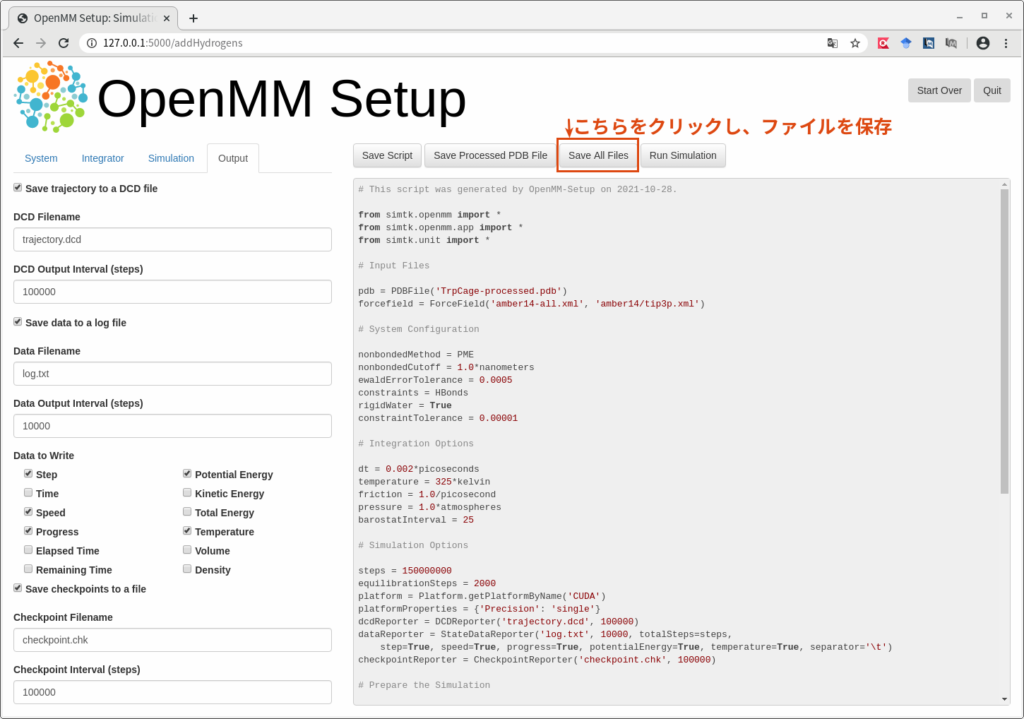
最後に、上のようにOutput を設定し、Save All Files をクリックし、ファイルを保存します(ダウンロードされます)。これで設定が完了です。
OpenMM によるシミュレーションの実行
上記設定が完了すると、openmm_simulation.zip ファイルが得られていますので、これを解凍します。openmm_simulation というフォルダが得られますので、そのなかで計算を実行します。
$ cd openmm_simulation
openmm_simulation のなかには、TrpCage.pdb、TrpCage-processed.pdb、 run_openmm_simulation.py という3つのファイルが得られています。TrpCage.pdbは前回構築した構造です。TrpCage-processed.pdb の方をPyMOLで開いて確認してみましょう。
$ pymol TrpCage-processed.pdb
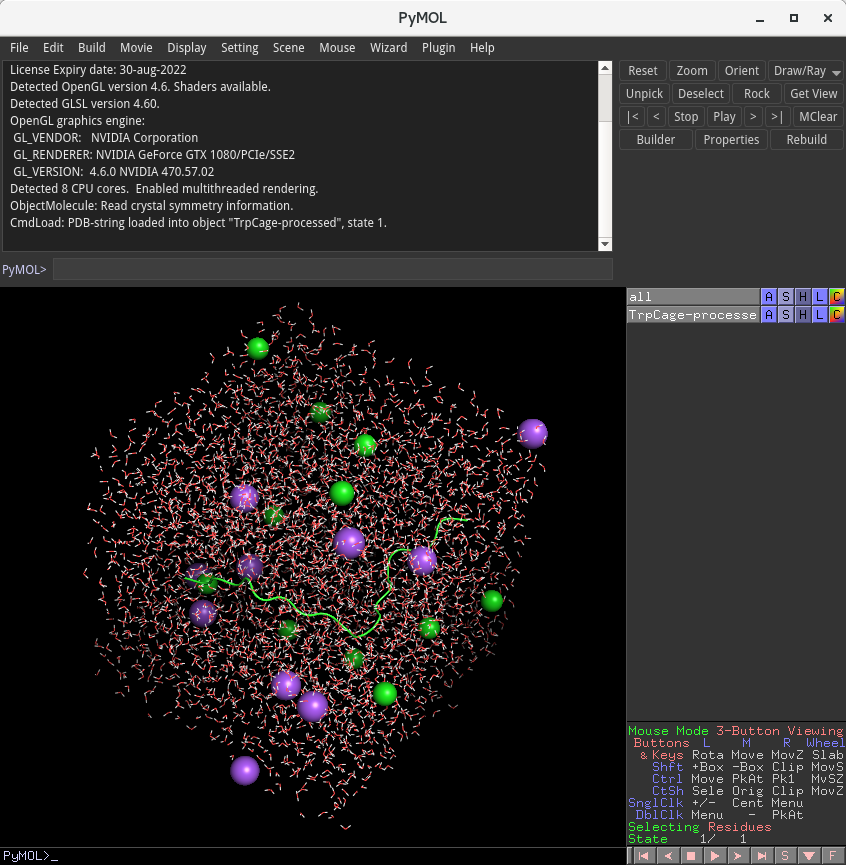
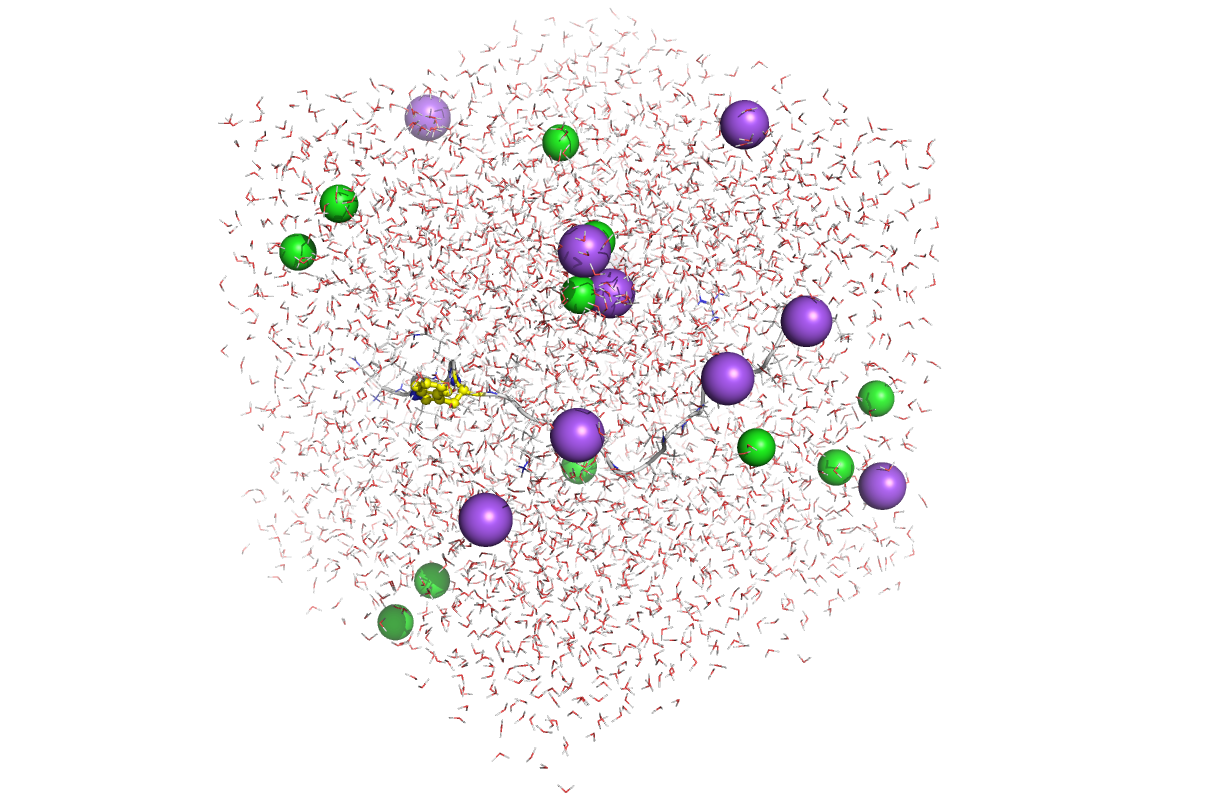
上記のように表示されました。Trp-cageの構造が緑色のリボンで、その周りに赤と白で示された水分子(H2O)が配置されています。紫および緑の球体は、Na+とCl– です。 これがシミュレーション boxの初期配置になります。
では、PyMOLを終了し、実際に計算を実行してみます。計算の設定が、run_openmm_simulation.py に記載されており、以下のコマンドを実行すれば、計算がスタートします。
$ python run_openmm_simulation.py
Warning: importing 'simtk.openmm' is deprecated. Import 'openmm' instead. Building system... Performing energy minimization... Equilibrating... Simulating...
すぐに上のような出力が得られ、計算が進みます。計算中、3つの出力ファイルが更新されていきます。
今回の記事では、OpenMM-Setup によるシミュレーションの設定と、OpenMMによる分子動力学計算を実行しました。シミュレーション結果については、次以降の記事で確認したいと思います。
-120x68.png)
コメント