
このカテゴリーの記事では、「Pythonを使った機械学習やケモ・バイオインフォマティクスの実装や論文」を紹介していきたいと考えています。Python は3系(3.7)、anacondaを中心にして環境構築していきます。以下のようなハード・ソフト環境(CentOS Linux)を用いますが、Python(anaconda)が利用できれば、異なる環境下でも類似の実装が可能かと思います。
● CPU Intel® Core™ i7-6700K CPU @ 4.00GHz × 8
● メモリ 64GB
● GPU GeForce GTX 1080/PCIe/SSE2
● OS CentOS Linux 8
● Python 3.7
はじめに
以前、タンパク質と低分子化合物のドッキングシミュレーション用ソフトウェアの一つである AutoDock Vina のインストールを紹介しました。
●AutoDock Vina 論文: O. Trott and A. J. Olson. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry, 31(2), 455-461.
●バージョンアップ(1.2.0)論文:J. Eberhardt, D. Santos-Martins, A. F. Tillack, and S. Forli. (2021). AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. Journal of Chemical Information and Modeling.
今回の記事では、AutoDock Vina のチュートリアルを実施してみたいと思います。さらに、結果の表示についても紹介したいと思います。
また、タンパク質-タンパク質間、タンパク質-ペプチド間、タンパク質-DNA間のドッキングシミュレーションのソフトウェア “LightDock” についての記事もアップしていますので、下記を参考にしてください。
Nglview のインストール
今回、ドッキングシミュレーション結果の表示に、Nglview を使ってみたいと思います。Nglview を使うことで、分子構造や分子動力学計算結果をJupyter notebook 上でインタラクティブに表示することが可能です。まずは、インストールしましょう。conda を利用して簡単に導入できます。
前回の記事でAutoDock Vina 1.2.2 をインストールしていれば、その仮想環境 vina をアクティブにしましょう。
$ conda activate vina # AutoDock Vina, Jupyter notebookをインストール済み仮想環境vinaをアクティブに
そのうえで、以下のコマンドを実行します。
$ conda install nglview -c conda-forge
この記事を書いている時点では、nglview 3.0.3 がインストールされました。
AutoDock Vinaのテストデータのダウンロード
こちらのページに記載されている通り、チュートリアルを実行してみたいと思います。まずは、以下のコマンドで、テストデータをダウンロードします。
$ wget https://raw.githubusercontent.com/ccsb-scripps/AutoDock-Vina/develop/example/python_scripting/1iep_receptor.pdbqt $ wget https://raw.githubusercontent.com/ccsb-scripps/AutoDock-Vina/develop/example/python_scripting/1iep_ligand.pdbqt
1行目で、タンパク質(receptor)である1iep_receptor.pdbqtを、2行目でリガンド(ligand)分子1iep_ligand.pdbqtのファイルをダウンロードしています。このテストデータは、Protein Data Bank (PDB)にてID:1IEPで登録されている、チロシンキナーゼとその阻害剤イマチニブの複合体構造です。チロシンキナーゼがreceptor、阻害剤イマチニブがligand です。今回、両者の複合体をドッキングシミュレーションしますが、実験的に決定された正解の複合体構造が、1IEP で登録されているわけです。構造の詳細は、こちらのリンクを参照してください。
また、イマチニブは、慢性骨髄性白血病 、KIT (CD117) 陽性消化管間質腫瘍 (GIST) に対する治療薬として用いられる、いわゆる分子標的抗がん剤の1種です。
AutoDock Vinaの実行
それでは、実際にチュートリアルを実行していきます。Jupyter notebookを起動します。
$ jupyter notebook
まずは、セルに以下を入力し、Run して下さい。
from vina import Vina
Python パッケージ vina が呼び出されました。
続いて、下記を実行します。
v = Vina(sf_name='vina')
この行では、Vinaオブジェクトを v として作成しています。分子のドッキングに使用される力場(スコアリング関数)も指定しています。ここでは Vina 力場を設定しました。AutoDock4 や Vinardo のような別の力場を使用したい場合は、vina を ad4 または vinardo に置き換えてください。
続いて、以下を実行してください。
v.set_receptor('1iep_receptor.pdbqt')
v.set_ligand_from_file('1iep_ligand.pdbqt')
v.compute_vina_maps(center=[15.190, 53.903, 16.917], box_size=[20, 20, 20])
ここでは、1行目で受容体 1iep_receptor.pdbqt というPDBQTファイルをロードしています。次の行は、1iep_ligand.pdbqt というリガンドファイルを読み込んでいます。3行目で、compute_vina_maps()関数によって、Vina 力場に基づいて各リガンドの原子タイプに対する親和性マップを計算しています。center と box_sizeで親和性マップを計算する領域を指定しています。受容体のこの領域にドッキングシミュレーションが実行されることになります。
最後に、以下でドッキングを実行することができます。
v.dock(exhaustiveness=32, n_poses=20)
v.write_poses('1iep_ligand_vina_out.pdbqt', n_poses=5, overwrite=True)
ここでは、exhaustiveness引数を指定し、32回のモンテカルロ・サンプリングを実行し、探索中に20個のポーズ(n_poses)を保存しています。最後に、1iep_ligand_vina_out.pdbqtというPDBQTファイルに、最上位の5つのポーズ(n_poses)を書き込んでいます。これらの値を変更することで、より多くのドッキングポーズを保存することが可能です。
ドッキング計算自体は、数分間で終了するかと思います。
それでは、Nglview を用いて、Jupyter notebook 上でドッキング構造を表示してみたいと思います。(*ドッキングのスコア(affinity, kcal/mol)は、1iep_ligand_vina_out.pdbqtをテキストエディタで開くことで確認できます。こちらも参考にしてください。)
以下でNglviewをインポートします。
import nglview
from IPython.display import display, Image
次に以下を実行します。
view = nglview.NGLWidget()
これでview オブジェクトが作成されました。これに対して、以下のようにドッキング結果(1iep_ligand_vina_out.pdbqt、ドッキング後のリガンド分子構造)および、受容体構造(1iep_receptor.pdbqt)を読み込み、表示します。
v1 = view.add_component("1iep_ligand_vina_out.pdbqt")
v1.clear()
v1.add_representation(repr_type='spacefill', selection='not hydrogen')
v2 = view.add_component("1iep_receptor.pdbqt")
v2.clear()
v2.add_representation(repr_type='cartoon', colorScheme = 'sstruc')
view._remote_call('setSize', target='Widget', args=['','600px'])
view
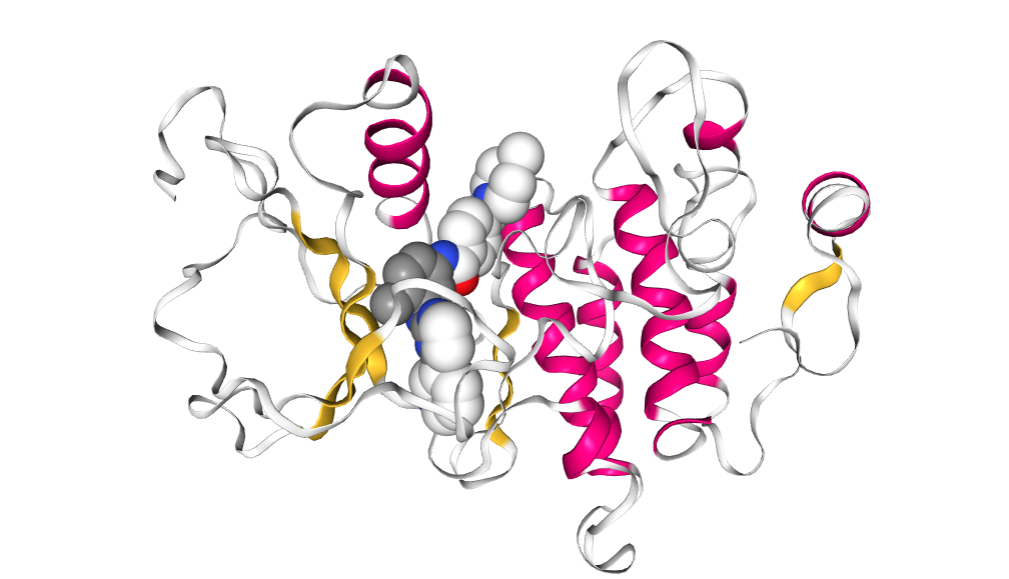
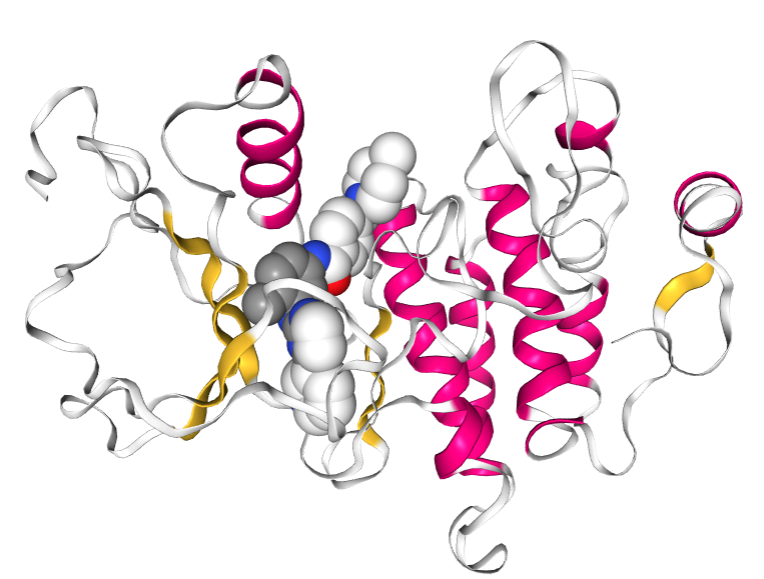
ドッキング結果のトップスコアを示した構造が表示されます。リガンド分子は空間充填モデルで、受容体はリボンモデルで表示しています。マウスで回転や拡大ができますので、確認してみてください。
ドッキング結果は、Pymolなど、他のソフトウェアで表示し、詳細に分析することが可能です。下記の記事も参照してください。

コメント