

このカテゴリーの記事では、「Pythonを使った機械学習やケモ・バイオインフォマティクスの実装や論文」を紹介していきたいと考えています。Python は3系(3.7)、anacondaを中心にして環境構築していきます。以下のようなハード・ソフト環境(CentOS Linux)を用いますが、Python(anaconda)が利用できれば、異なる環境下でも類似の実装が可能かと思います。
● CPU Intel® Core™ i7-6700K CPU @ 4.00GHz × 8
● メモリ 64GB
● GPU GeForce GTX 1080/PCIe/SSE2
● OS CentOS Linux 8
● Python 3.7
はじめに
これまで、以下のような記事で、機械学習的手法の実装として DeepChem を、さらに、タンパク質のドッキングシミュレーションの実践として AutoDock Vina と LightDock を紹介してきました。
ドッキングシミュレーションでは、タンパク質と他の分子の構造情報を用いて、それらの結合をシミュレーションします。多くの場合、分子間の相互作用は、古典的な(非量子化学的な)近似のもとでスコア化して計算され、推定される結合(結合エネルギー)の正確さは低いものとなります。また通常、分子構造はある1つの状態として扱われ、分子のランダムな熱運動や秩序あるゆらぎ、大きな構造変化等は考慮されません。
また、機械学習的手法においては、あらわに分子間相互作用を計算するのではなく、多くの相互作用データを、柔軟なモデルで学習し、その学習済みモデルで未知の相互作用を予測します。この点、決まったモデルを基に相互作用を計算するドッキングシミュレーションとは、対象的です。また本手法では、事前に予測目的に応じた、いわゆるビッグデータ(分子間相互作用予測の場合には、タンパク質がどのような分子とどの程度の強さで結合するかのデータ)が必要です。さらに、特に深層学習においては、そのモデルが非常に柔軟である替わりに、得られた予測結果の要因を解釈することが困難である場合が多いです。
機械学習的手法はケモ・バイオインフォマティクスの代表例ですが、ドッキングシミュレーションは、計算科学(計算化学)的手法と呼ばれることが多いと思います。
このような計算科学的な手法として、他には「分子動力学法(Molecular dynamics)」がよく用いられます。分子動力学法(Molecular dynamics)は、原子・分子の物理的な「動き」をシミュレーションする手法です。従って、分子のゆらぎや構造変化を考慮した相互作用の計算・予測が可能です。また通常、分子の動きを計算するためのポテンシャル関数(力場)は古典的なものが用いられますが、局所的に(計算時間を考慮しなければ、全体的に)量子力学に基く力場を利用することも可能です。これらのことから、一般的にドッキングと比較して、より精度の高いシミュレーション法であると考えられます。
そこで本記事より、分子動力学法(Molecular dynamics)の実装として、OpenMM を紹介していきたいと思います。 論文は下記を参照ください。
●論文 P. Eastman, J. Swails, J. D. Chodera, R. T. McGibbon, Y. Zhao, K. A. Beauchamp, L.-P. Wang, A. C. Simmonett, M. P. Harrigan, C. D. Stern, R. P. Wiewiora, B. R. Brooks, and V. S. Pande. “OpenMM 7: Rapid development of high performance algorithms for molecular dynamics.” PLOS Comp. Biol. 13(7): e1005659. (2017)
分子動力学計算を実行するためのソフトウェアとして、Amber、CHARMM、NAMD、GROMACS 等が有名です。また、既存の力場としては、Amber、CHARMM、OPLS等が広く用いられています(*Amber、CHARMMは、計算を実行するソフトウェアを指す場合と、力場を指す場合があります。)。これらは、それぞれに実績のある優秀なものですが、ソフトウェアの導入には、一定の知識や手間が必要です。特に近年、分子動力学計算においてもGPUを用いた計算の並列化・高速化が求められていますが、これにも少し慣れが必要です。
一方、OpenMMはCondaを利用して簡単に導入することができ、GPU利用環境も同時に構築できます。以下で実際にインストールしてみましょう。
OpenMM のインストール
まずは下記のように仮想環境 openmmを用意して、アクティブにしましょう。
$ mkdir OpenMM #OpenMMディレクトリを作成 $ cd OpenMM #OpenMMディレクトリに移動 $ conda create -n openmm python=3.8 #python 3.8 の仮想環境openmm を作成 $ conda activate openmm #仮想環境openmm をアクティブに
この環境下に conda で OpenMM をインストールします。こちらのサイトの内容も参照してください。実際には、以下のコマンドを実行するだけです。
$ conda install -c conda-forge openmm cudatoolkit=11.2
これだけで、インストールが完了します。2021年10月の記事時点では、OpenMM 7.6 がインストールされました。また、OpenMMと同時に CUDA Toolkit もインストールしました。CUDA Toolkitは、今回のようにNVIDIA社製GPU を数値計算用途で使うために必要となるソフトウェアライブラリです。
さらに、そのまま以下を実行します。
$ python -m openmm.testInstallation
OpenMM Version: 7.6 Git Revision: ad113a0cb37991a2de67a08026cf3b91616bafbe There are 4 Platforms available: 1 Reference - Successfully computed forces 2 CPU - Successfully computed forces 3 CUDA - Successfully computed forces 4 OpenCL - Successfully computed forces Median difference in forces between platforms: Reference vs. CPU: 6.29511e-06 Reference vs. CUDA: 6.73294e-06 CPU vs. CUDA: 7.36796e-07 Reference vs. OpenCL: 6.76294e-06 CPU vs. OpenCL: 8.07109e-07 CUDA vs. OpenCL: 2.73444e-07 All differences are within tolerance.
上のような出力が得られれば、インストールが完了しています。本サイトでは、Cent OS Linux のPCを用いて実装を行っていますが、Windows やMAC でも、conda を利用するだけで簡単にインストールすることができるはずです。
anaconda および conda について、Web上に多くの情報がありますが、以下の記事も参考にして下さい。

また、NVIDIA のGPUを搭載したPC であれば、CUDA Toolkitの利用で、GPUを利用した計算が可能ですが、事前にGPUドライバーのインストールは必要になります。本家サイト等を参考にしてください。
OpenMMによる分子動力学計算の実行:はじめの一歩
OpenMMでは、こちらのページのような examples を用意してくれています。詳細はあとにして、とりあえず計算を実行してみたいと思います。最初の例題として、simulatePdb.py ファイルとinput.pdb ファイルを使います。例題に必要なファイルは、本記事と同様にanaconda3 で環境構築している場合、anaconda3/envs/openmm/share/openmm/examples/ に作成されています。以下のコマンドでコピーします。xxxxにはホームディレクトリ名を入れてください。
$ cp /home/xxxx/anaconda3/envs/openmm/share/openmm/examples/simulatePdb.py ./ $ cp /home/xxxx/anaconda3/envs/openmm/share/openmm/examples/input.pdb ./
あとは、下記を実行します。今コピーしたファイルを指定する形になります。
$ python simulatePdb.py
#"Step","Potential Energy (kJ/mole)","Temperature (K)" 1000,-142175.18593246874,290.62817367197846 2000,-140534.18593246874,301.851677950669 3000,-140688.49843246874,303.33218564964955 4000,-140854.87343246874,300.78528393866435 5000,-140908.49843246874,300.5157975221865 6000,-140554.77968246874,306.404194729315 7000,-140822.56093246874,301.43576875557653 8000,-141083.65468246874,300.89589254952153 9000,-140411.49843246874,301.28868144804585 10000,-140783.12343246874,297.618784360445
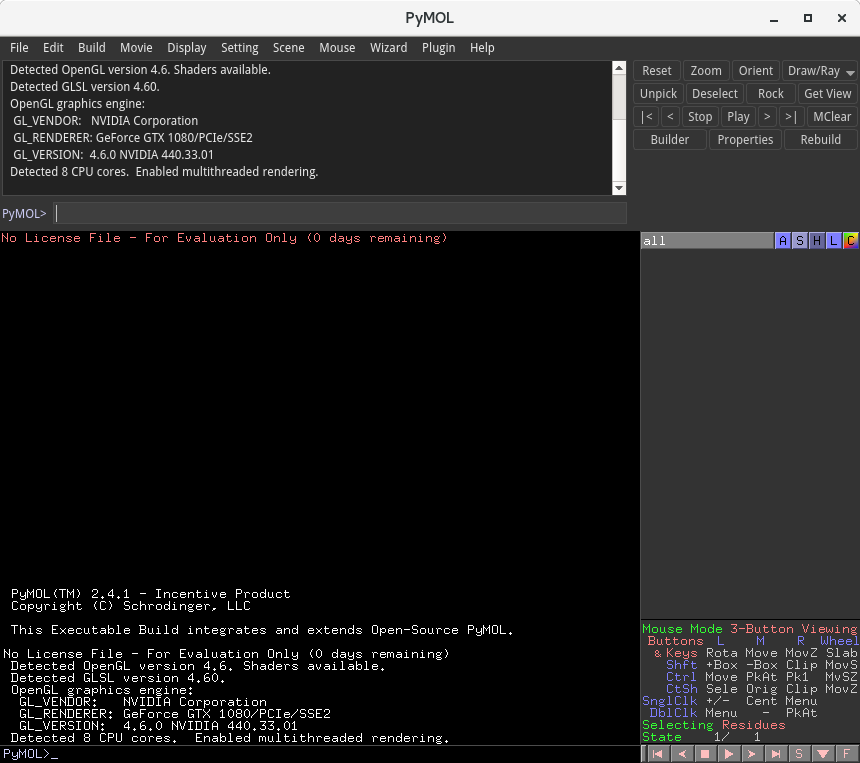
1分足らずのうちに、上記のような計算結果が段階的に出力されたと思います。これで、10,000 ステップの計算が完了しています。また、output.pdb ファイルが作成されているかと思います。このファイルに、上記計算結果が出力されており、PyMOLなどの分子表示ソフトウェアで開くことで、結果を確認できます。PyMOLについては、以下の記事も参考にしてください。
以下を実行して、この仮想環境にPyMOLをインストールしておきましょう。
$ conda install -c schrodinger pymol
インストールできたら、以下を実行して、PyMOLでoutput.pdb ファイルを開いてみましょう。
$ pymol output.pdb
PyMOLウインドウが起動し、以下のように表示されるはずです。
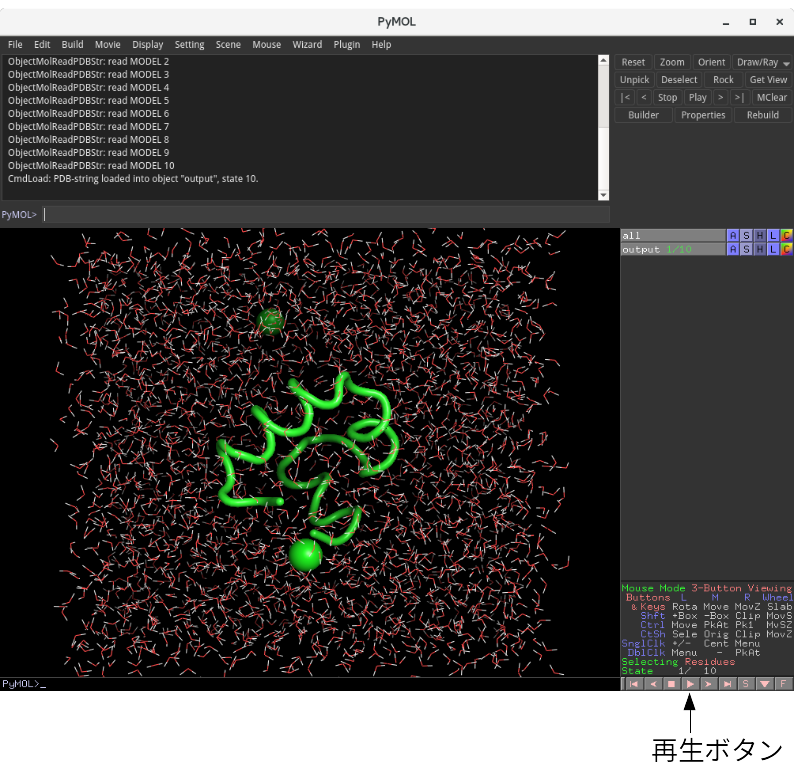
緑のひもがタンパク質分子を、緑の球が塩化物イオンを、赤と白の線が水分子を表しています。この例題では、22個のアミノ酸でできた小さなタンパク質について、水中での挙動が水分子の運動も考慮した形でシミュレーションされています。
右下に配置されている再生ボタン▶︎を押してみてください。
以下のように、分子動力学計算結果である分子の「動き」が表示されたかと思います。
動画をクリックして下さい。
今回、分子動力学法(Molecular dynamics)シミュレーションを取り上げ、その実装としてOpenMMを紹介しました。インストールと例題の実行が簡単にできました。次回以降、計算の詳細や他の例題について紹介していきたいと思います。
コメント