

1. 研究の背景と目的
医薬品候補を設計する際、ターゲットとなるタンパク質や医薬候補リガンドの立体構造情報をベースにする手法を、Structure Based Drug Design(SBDD)と呼びます。ウェットな実験が当然必要となりますが、コンピュータ上で「リガンドはタンパク質のどこに、どのように結合するのか?」(構造予測)に加えて、「その結合はどれほど強いのか?」(結合親和性予測)が推定できるのであれば、SBDDの効率が上がると考えられます。
AlphaFold21の登場、さらにはAlphaFold32やBoltz-13といった後継モデルによって、タンパク質のみならず、複数のリガンドとの複合体構造についても高精度な予測が可能になっています。しかし、正確な予測構造が得られても、創薬判断に足る精度で結合親和性を推定するには、計算の精度と速度において、トレードオフが存在しました。
・物理ベースシミュレーション (FEP): タンパク質-リガンド間の結合自由エネルギー変化(ΔG)、結合親和性を第一原理的に算出する方法です。実験値に匹敵する精度があるとされますが、1つの化合物評価に数日を要します。
・分子ドッキング: タンパク質-リガンドの適合度をスコアとして評価する方法で、高速にスクリーニングできます。しかし、そのスコアは親和性を正確には反映しておらず、精度は限定的です。
Boltz-2 は、タンパク質-リガンドの構造予測と結合親和性予測を同時に実行できるAIモデルであり、FEPに匹敵する精度と、ドッキングに迫る速度(FEP比で1000倍以上高速)を実現した!! とのことです。スゴイ時代!
Boltz-2 paper: Passaro et al., (2025) Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction, bioRxiv 2025.06.14.659707; doi: 10.1101/2025.06.14.659707
Boltz-2 Github: Boltz-2 https://github.com/jwohlwend/boltz
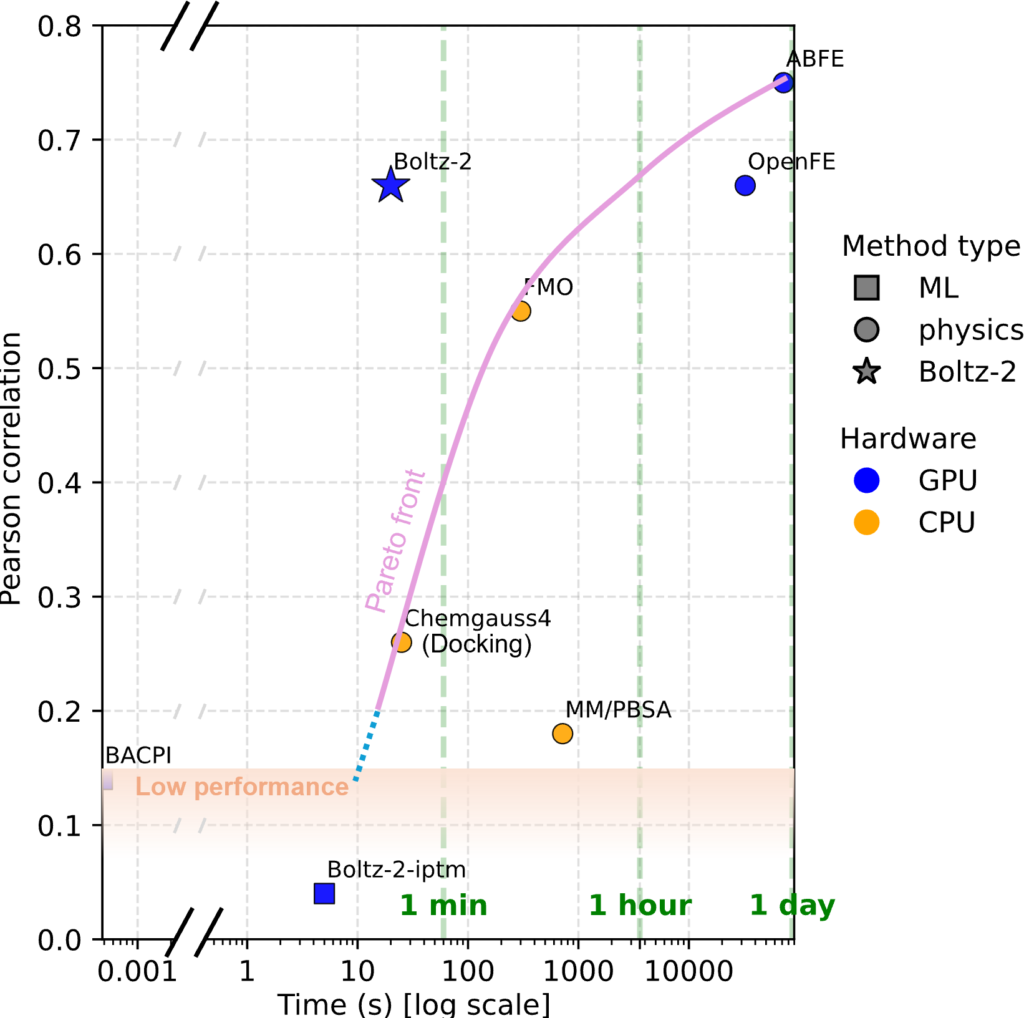
Fig. 1: 図1: Boltz-2は親和性予測において精度と速度のトレードオフを強く示す。プロットは、タンパク質-リガンドベンチマークの4ターゲットサブセット(CDK2, TYK2, JNK1, p38)に基づく。
引用 Boltz-2 paper
この記事では、Boltz-2の論文とコード、特に結合親和性予測のAffinity Moduleについて勉強しました。
2. Bolts-2のメソッド、リザルト、および関連コード
Boltz-2は、Boltz-1類似の構造予測に加えて、結合親和性を学習できるアーキテクチャになっています。

Fig. 2: Boltz-2モデルアーキテクチャ図。
引用 Boltz-2 paper
Boltz-2は、4つの主要コンポーネントからなり、それらがFig.2の関係性で機能します。Boltz-1には無かった、Confidence ModuleとAffinity Moduleが、TrunkとDenoising Moduleの後段に付いており、構造表現と情報を利用して親和性を予測します。
Ⅰ. 構造表現コンポーネント
・Trunk :シーケンス情報やMSAから、分子間および分子内のペアワイズ表現(ztrunk)を構築します(PairFormerモジュール群を使用)。
Ⅱ. 構造生成コンポーネント
・Denoising Module:3D座標の生成・精緻化を担う拡散モデルベースのモジュール。Trunkからの構造表現をアテンションバイアスなどの形で受け取り、複合体の3D原子座標(xpred)を生成します。
Ⅲ. 後続の分析・予測モジュール
Affinity ModuleとConfidence Moduleは、Trunkが学習した相互作用の表現(最終的なペア表現(ztrunk)と入力単一表現 (sinputs))とDenoising Moduleによって予測が完了した3D構造(トークン座標のディストグラム)の両方を入力として利用し、機能します。
・Affinity Module : 内部で専用のPairFormerを用い、タンパク質—リガンドおよびリガンド内の相互作用のみに焦点を絞って処理を行い、最終的に結合の有無を判定するための結合尤度(二値分類)と親和性値(連続値回帰)を出力します。
・Confidence Module : 予測された構造の信頼度を評価します。Affinity Moduleと同様にTrunkとDenoising Moduleより前述の表現情報と3D構造情報をうけとり、予測構造の品質(LDDTやPAEなど)を評価します。
2.1 Boltz-2の核心(1): 結合親和性予測モジュール (Affinity Module)
Boltz-2がFEPに迫る精度を達成できた核心は、Affinity Moduleにあります 。Affinity Moduleの機能は、大きく分けて「特徴量の準備・強化」と「相互作用に基づく予測」の2つの処理で構成されています。
フェーズ I: 特徴量の準備・強化 (Feature Preparation and Refinement)
1. 座標情報からの特徴量抽出(ディストグラムの計算)
Trunkが生成したペア表現・単一表現をもとにDenoising Moduleで生成した3D原子座標(x_pred)を受け取り、トークン間の距離情報(ディストグラム)を生成します。予測した複合体構造を定量的に理解することが狙いです。
AffinityModuleのforwardメソッド内では、まず代表原子の座標を抽出し、それらの間の距離を計算します。
x_pred_repr = torch.bmm(token_to_rep_atom.float(), x_pred)
# 座標間の距離を計算
d = torch.cdist(x_pred_repr, x_pred_repr)
# 距離をビニングしてディストグラムを生成し、埋め込みを行う
distogram = (d.unsqueeze(-1) > self.boundaries).sum(dim=-1).long()
distogram = self.dist_bin_pairwise_embed(distogram)このディストグラム(distogram)は、Pairwise Conditioningを通じて、Trunkから引き継がれたペア表現(z)に結合情報として組み込まれ、表現を強化します。
z = z + self.pairwise_conditioner(z_trunk=z, token_rel_pos_feats=distogram)このzは次の2.の工程で使用されます。
2. タンパク質-リガンド相互作用に特化したマスキング
Affinity Moduleでは、親和性予測の焦点は結合界面に限定されるように設計されています。つまり、複合体構造全体の相互作用ではなく、リガンドと結合ポケット周辺、およびリガンド分子内部の相互作用のみをPairFormerに処理させています。
この選択的な処理は、cross_pair_maskの作成によって実現されています。これは、タンパク質トークン(rec_mask)とリガンドトークン(lig_mask)のインデックス情報を用いて構築されます。
# cross_pair_maskの定義の一部
rec_mask = (feats["mol_type"] == 0).repeat_interleave(multiplicity, 0)
# mol_type == 0 はタンパク質(受容体)を示す
lig_mask = (
feats["affinity_token_mask"]
.repeat_interleave(multiplicity, 0)
.to(torch.bool)
)
# affinity_token_mask はリガンドを示す
cross_pair_mask = (
lig_mask[:, :, None] * rec_mask[:, None, :] # リガンド-タンパク質
+ rec_mask[:, :, None] * lig_mask[:, None, :] # タンパク質-リガンド
+ lig_mask[:, :, None] * lig_mask[:, None, :] # リガンド-リガンド(自己結合を含む)
)この cross_pair_mask が Pairformer モジュールに渡され、指定された相互作用のみが計算対象となります。
z = self.pairformer_stack(
z, # 入力:Trunk情報とディストグラムを含むペア表現
pair_mask=cross_pair_mask, # 適用:タンパク質-タンパク質相互作用を排除
use_kernels=use_kernels,
)PairFormer Stack(self.pairformer_stack)は、複数のPairFormer層(モデルによって4層または8層)を積み重ねたものです。これによって、入力されたペア表現 z が、結合親和性予測に特化したより強力で洗練された相互作用特徴量へと反復的に更新されます。
フェーズ II: 相互作用に基づく予測 (Interaction-Based Prediction)
3. グローバルプーリングと専用予測ヘッド
PairFormerで処理された相互作用ペア表現(z)は、最終的にグローバルプーリング(平均化)によって単一のコンテキストベクトル(g)に集約されます。
# cross_pair_maskでマスクされた領域の平均プーリングを実行
g = torch.sum(z * cross_pair_mask, dim=(1, 2)) / (
torch.sum(cross_pair_mask, dim=(1, 2)) + 1e-7
)この集約されたコンテキストベクトルgは、AffinityHeadsTransformer内の2つの独立したMLP(多層パーセプトロン)ヘッドに入力され、それぞれの予測値が出力されます。
■結合親和性(連続値回帰): IC50などの結合強度を示す連続値を予測します。
# 結合親和性を求める専用ヘッドの定義 (ReLUと線形層のMLP)
self.to_affinity_pred_value = nn.Sequential(
nn.Linear(input_token_s, input_token_s),
nn.ReLU(),
nn.Linear(input_token_s, input_token_s),
nn.ReLU(),
nn.Linear(input_token_s, 1),
)
# 最終的な出力値
affinity_pred_value = self.to_affinity_pred_value(g).reshape(-1, 1)■結合尤度(二値分類): 結合の有無を判定するための二値分類のロジットを出力します。
# 結合尤度を求めるためのスコアヘッドの定義:
self.to_affinity_pred_score = nn.Sequential(
nn.Linear(input_token_s, input_token_s),
nn.ReLU(),
nn.Linear(input_token_s, input_token_s),
nn.ReLU(),
nn.Linear(input_token_s, 1),
)
self.to_affinity_logits_binary = nn.Linear(1, 1)
# 最終的な出力値
affinity_pred_score = self.to_affinity_pred_score(g).reshape(-1, 1)
affinity_logits_binary = self.to_affinity_logits_binary(affinity_pred_score).reshape(-1, 1)これらの専用ヘッドが、ヒット探索に必要な結合尤度(二値分類)と、リード化合物の最適化に必要な親和性値(連続値)という、異なる情報を出力します。最後はMLPに入力するだけ…!!?
この設計により、Boltz-2はリード最適化ベンチマーク(FEP+やOpenFE)において、従来の機械学習やドッキング手法を圧倒し、計算コストが1000倍以上かかるFEP法に肉薄する予測精度(ピアソン相関係数 R≈0.62〜0.66)を達成しました。さらに、CASP16の親和性予測タスクで、事後評価ながら公式参加の全チームを凌駕する性能を示しています(Fig. 6)。
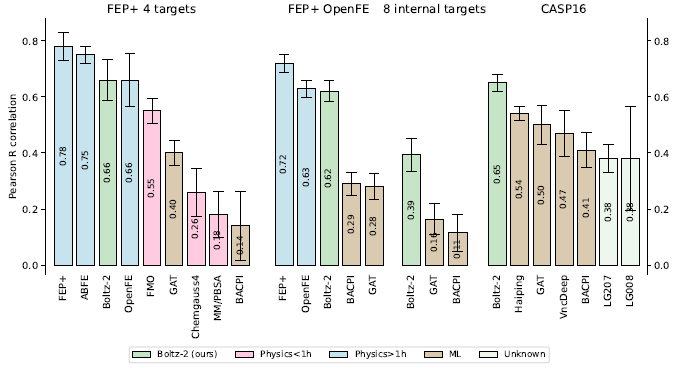
Fig. 6:4つの親和性値テストセットにおける各アッセイの平均ピアソン相関係数。
引用 Boltz-2 paper
2.2 Boltz-2の核心(2): 「構造品質フィルター」によるデータ戦略
高精度な親和性予測モデルの構築には、アーキテクチャ以上に良質な学習用データが不可欠です。著者らは、公開DBの親和性データの品質の問題に対して、以下のように対処しています。
1. 親和性値の標準化と化学的ノイズ対策
データソースは、主に連続的な親和性測定値(Ki, Kd, IC50など)を提供するChEMBL、BindingDB、および二値分類データを提供するPubChem HTS、CeMM Fragment Dataset、MIDAS Metabolite Dataが使用されました。すべての連続的な親和性値は、μMを基準としたlog10スケールに変換されています。PAINS(Pan-Assay Interference Compounds)化合物の除去や、重原子が50を超える大きなリガンドの除外など、化学的ノイズ源となる化合物のフィルタリングが実施されました。
2. 構造品質フィルターによるデータ選別
1.で得られたデータについて、Boltz-2の構造予測モデル(Trunk)でランダムに選ばれたバインダー10個の複合体構造をBoltz-2の確信度モジュールで予測し、その平均予測界面pTM(ipTM)スコアが0.75を超えるアッセイのみを学習データに残しています。
3. ペアワイズ差分損失による学習シグナルの強調
学習時に、実験間のばらつきの影響を打ち消し、真の活性の差をモデルに強く学習させるため、損失関数に重み付けがされています。具体的には、同一の実験報告内の類似化合物ペアの親和性値の差分(Ldif)を学習する項に、大きな重み(0.9)を持たせています。
2.3 Boltz-2の核心 (3): 仮説検証を可能にする「制御可能性 (Controllability)」
Boltz-2は、既存の実験的証拠や何らかの仮説を、構造予測プロセスに組み込むことができる設計となっています。この機能は、構造予測を担うDenoising Moduleの推論プロセスに、ユーザー定義の制約をポテンシャル関数として適用するBoltz-steeringです。
Steering技術の仕組み
Boltz-steeringは、Boltz-1x3で導入された、物理的な不正確さ(ステレオ化学の誤りや立体衝突など)を修正する技術を統合・拡張したものです。これは、Feynmac-Kac (FK) ステアリングフレームワークに基づいて構築されており、 AtomDiffusion.sampleメソッド内で以下の手順で実行されます。
1. ポテンシャル関数の利用: モデルは get_potentials(steering_args, boltz2=True) を呼び出し、ユーザーが指定した条件(テンプレート、コンタクトなど)や物理法則をエネルギーポテンシャルとして定義します。
2. Feynmac-Kac (FK) ステアリング: 推論中の拡散プロセス(逆拡散)の各中間ステップで、ポテンシャルエネルギーの低い経路にサンプリング軌道を傾けるリサンプリングが行われます。
3. 勾配誘導 (Guidance): 推論プロセス中に、ポテンシャルエネルギーを最小化するように、予測座標に勾配降下法による更新が適用されます。physical_guidance_update やcontact_guidance_update が有効な場合、この勾配に基づく更新が実行されます。
3つの主要な制御可能性
・実験手法の条件付け: X線結晶構造解析、NMR、MDなど、構造データの元となった実験手法を指定できます。これにより、予測結果を特定の実験手法で得られる構造アンサンブルに近づけることが可能になります
・テンプレートの条件付けとステアリング: 構造が類似する複合体の既知の構造(テンプレート)を予測に利用します。Boltz-1にはなかった機能で、AlphaFold3と異なりマルチ鎖(多量体)テンプレートをサポートします。ユーザーは、ソフトな条件付けだけでなく、ステアリングポテンシャルを用いて、予測された構造がテンプレートの特定の領域から閾値(αcutoff Å)以内の距離になるよう、モデルに厳密に強制することができます。
・接触・ポケットの条件付けとステアリング:実験的知見や仮説に基づき、トークン(残基や原子)間に特定の距離制約(閾値 rAB )を課します。これらの制約は、物理ポテンシャル(ステアリングポテンシャル)として推論時の拡散プロセスに組み込まれ、予測構造を厳密に誘導する機能を提供します。
3. 発展的な使用例:予測から「生成」へ (TYK2ターゲット)
Boltz-2の発展的な使用例として、高速かつ高精度なスコアリング関数として機能し、分子生成モデルと連携できる点にあります。
・論文での実証(TYK2ターゲット): キナーゼ阻害剤のターゲットであるTYK2に対し、分子生成モデル(SynFlowNet5)と組み合わせたバーチャルスクリーニングが実行されています。SynFlowNetは、GFlowNet をベースとし、化学反応とビルディングブロック空間で分子を設計することで、生成される分子の合成可能性を保証するモデルです。Boltz-2の予測スコアを報酬としてSynFlowNetを学習させた結果 、既存の化合物ライブラリ(Enamine HLL、46万化合物)を網羅的にスクリーニングするよりも少ない評価回数(11.7万回)で、より強力なリガンドを発見することに成功しました。
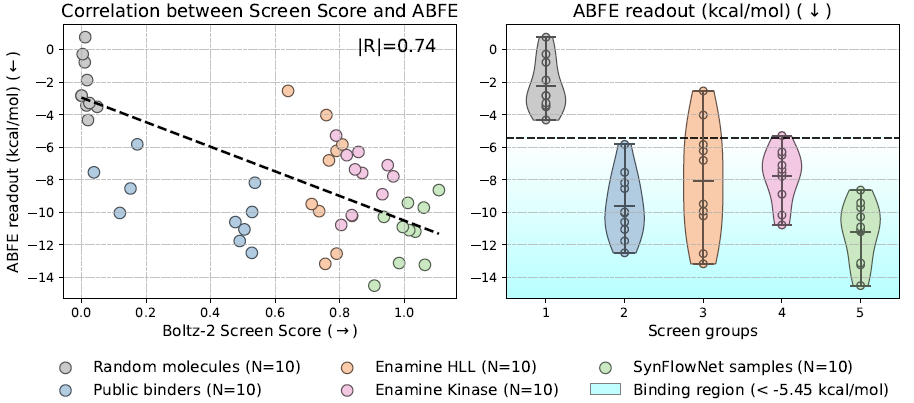
Fig.8: TYK2タンパク質に対して実施した仮想スクリーニング実験。左:各仮想スクリーニングストリームの最終化合物セットのBoltz-2スクリーニングスコアと絶対結合自由エネルギー(ABFE)推定値ΔGの相関(|R| = 0.74)。右:異なるスクリーニング戦略により提案された化合物のABFE予測ΔG分布。
引用 Boltz-2 paper
さらに、生成された分子は既知のTYK2阻害剤との構造類似性が低く、この手法が広い化学空間を探索する能力を持つことが推定されています。
4. まとめと将来展望
Boltz-2の「結合親和性予測」はまさに画期的です。様々な検証が必要ではありますが、学習データに含まれるタンパク質ーリガンドと類似する系であれば、かなりの精度が期待できるのではないでしょうか。CASP16に対する事後評価はそのことを十分に示唆していると考えられます。
一方で、親和性予測は構造予測の精度に依存し、タンパク質の大きな構造変化を伴うリガンド結合や、周囲の水分子やイオン強度を明示的に扱えない等、他の計算科学的手法や実験手法との連携も必要です。分子生成モデル(SynFlowNet)との組み合わせ例のように、Boltz-2をどのようなタスクに適用すると面白いのか考えてみたいと思います。
同じグループから、より最近、BoltzGenというタンパク質生成モデルも発表されています。こちらも非常に強力なツールのようです。

参考文献
1. Jumper, J. et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 doi: 10.1038/s41586-021-03819-2
2. Abramson, J. et al. (2024) Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500. doi: 10.1038/s41586-024-07487-w
3. Wohlwend, J. et al. (2025) Boltz-1 democratizing biomolecular interaction modeling. bioRxivdoi:10.1101/2024.11.19.624167.
4. Boitreaud J., et al. (2024) Chai-1: Decoding the molecular interactions of life. bioRxiv, doi: 10.1101/2024.10.10.615955.
5.Cretu, M., et al. (2024) SynFlowNet: Design of Diverse and Novel Molecules with Synthesis Constraints. arXiv:2405.01155, doi: 10.48550/arXiv.2405.01155

コメント