
1. 研究の背景と目的:単一配列モデルから検索拡張へ
近年、凄まじい性能を見せている、生成AIのChatGPTやGeminiなどは、いわゆる大規模言語モデル(LLM: Large Language Model)として開発されてきました。LLMをタンパク質に応用することを考えれば、「大規模な文章データ」は「多くのタンパク質のアミノ酸配列(一次構造)」ということになり、「単語(トークン)」は「アミノ酸」ということになります。こうして生み出されたのがタンパク質言語モデル(PLM)です。タンパク質の場合、近年の次世代シーケンス技術の発展で、様々な生物のゲノム情報が明らかとなり、タンパク質の一次構造情報が蓄積されている、即ち、「大規模な文章データ」に相当するデータが用意されています。スゴイ時代!!!
また、一次構造を大規模学習したモデルとは言えないかもですが、タンパク質立体構造予測 AlphaFoldのエンコーダ部のEvoformer(AlphaFold2)やPairformer(AlphaFold3)も、言語モデル的なモノです。
エンコーダ型のPLMとして、単一配列タンパク質言語モデルであるESM-21やProtTrans2などが代表的なモデルで、タンパク質を言語のように学習することで、タンパク質の構造的・機能的・進化的情報を獲得している(タンパク質とは何かを理解している!)と考えられ、後段のタスクに対して高い性能を示します。一方で、以下の「パラメータ・ボトルネック」が顕在化しています 。
1. パラメータの非効率性: 一次構造情報が有している、様々な情報(全般に共通する性質や、ある種のタンパク質に特異的な性質など)の全てをモデルのパラメータに圧縮しようとするため、モデルサイズが大きくなり、学習効率が悪い(*これは、良い面もあると思います。上手に圧縮できるなら、サイコー!?)。
2. 系統発生的バイアス: データの偏り(系統発生に起因するバイアスや、遺伝的浮動、ある生物種に偏ったサンプリングバイアス等)を学習してしまい、機能的に重要と誤認する、あるいは、低く評価してしまう。
これに対し、訓練時および推論時に、外部データベースから検索した相同配列(Multiple Sequence Alignments (MSA) や非整列相同配列セット)を明示的にモデルに入力提供する、「検索拡張PLM(Retrieval-Augmented PLM, RA-PLM)」が登場しています(MSA Transformer3やPoET4)。
本記事では、MSAを不要にする「アライメントフリー」なアーキテクチャと4兆トークン規模の学習を組み合わせ、単一配列モデルとRAモデルの最高性能(SOTA)を両立させた、Profluent-E1について勉強しました。Profluent-E1は、Profluent社(OpenCRISPR-1やProGen3などを発表)によって開発、2025年11月に公開されたモデルです。
Profluent-E1 paper: Jain, S. et al. (2025) E1: Retrieval-Augmented Protein Encoder Models. bioRxiv doi: 10.1101/2025.11.12.688125
Profluent-E1 GitHub: Profluent-E1 https://github.com/Profluent-AI/E1
2. 技術的核心とリザルト:アライメントフリーを実現するアーキテクチャと学習戦略
Profluent-E1の技術的核心は、「アライメントフリーを実現するAttention機構」と、それを支える「カリキュラム学習」の2点にあります。
2.1 Profluent-E1の核心①:アライメントフリーを実現するAttention機構
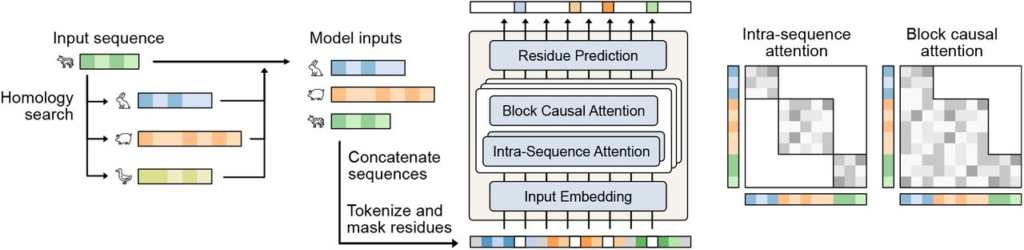
Fig. 1 Profluent-E1 Architecture
引用 Profluent-E1 paper
Profluent-E1は、クエリ配列の前に相同配列を連結したマルチシーケンス入力を使用し、この入力に対して**配列内アテンション (Intra-sequence Attention)とブロック因果アテンション (Block-causal Attention)**を交互に適用することで、進化的な文脈を効果的に学習します。
このメカニズムは、以下のステップで実現されています。
Step 1: 動的な入力構築と位置情報の生成 (src/E1/batch_preparer.py)
E1BatchPreparerクラスは、カンマ区切りで入力されたシーケンス(相同配列 + クエリ配列)を連結し、個々の配列の境界や相対位置、全体の絶対位置を識別できるように、以下の3種類のIDを付与します。
・within_seq_position_ids: 個々のタンパク質配列内における相対的な位置
・global_position_ids: 連結されたマルチシーケンス全体での絶対位置
・sequence_ids: 各トークンがどの配列(ホモログかクエリか)に属するか
# 1. within_seq_position_ids (各単一配列内の位置IDを結合)
within_seq_position_ids = torch.cat([encoding["position_ids"] for encoding in single_sequence_encodings])
# 2. global_position_ids (連結された全体の位置IDを計算)
global_position_ids, ctx_len = [], 0
for encoding in single_sequence_encodings:
global_position_ids.append(encoding["position_ids"] + ctx_len)
ctx_len = max(ctx_len, encoding["position_ids"].max().item() + ctx_len + 1)
global_position_ids = torch.cat(global_position_ids)
# 3. sequence_ids (各配列にIDを割り当て)
# num_tokensは各配列の長さのリスト
sequence_ids = torch.repeat_interleave(torch.tensor(num_tokens))上記のsequence_idsによって、モデルはどのトークンが「先行する相同配列」に属し、どのトークンが「クエリ配列」に属するかを区別できます。
Step 2: ブロック因果アテンションの切り替え (src/E1/model/attention.py)
Profluent-E1のTransformer層は、アテンションの種類を層ごとに切り替える「交互アテンションアーキテクチャ(alternating attention architecture)」を採用しています。
・Intra-sequence Attention (WITHIN_SEQ): トークンは自身の属する単一配列内の他のトークンにのみアテンションを適用します。
・Block-causal Attention (GLOBAL): トークンは自身の属する配列と、それに先行するすべての相同配列のトークンにアテンションを適用します。これが「検索拡張」の核となる層です。
この切り替えのロジックは、アテンション層のインデックス(layer_idx)に基づき、Attentionクラスの初期化時に決定されます。
# 層のタイプを決定
if self.config.global_attention_every_n_layers > 0:
self.layer_type = (
AttentionLayerType.GLOBAL
if (self.layer_idx + 1) % self.config.global_attention_every_n_layers == 0 # 特定の層でのみGLOBAL
else AttentionLayerType.WITHIN_SEQ
)
else:
self.layer_type = AttentionLayerType.WITHIN_SEQE1Configの設定に基づき、層のタイプが決定されます。これにより、適用される位置埋め込み(RoPE)のパラメータも動的に切り替えられます。
Step 3: 因果関係の強制とFlex Attention (src/E1/model/flex_attention.py)
Block-causal Attentionが適用される層では、Flex AttentionまたはFlash Attentionが使用されます。特に、学習時やキャッシュが埋まっていない推論時には、flex_attention_funcがブロック因果マスクを適用することで、因果性を強制します。
この因果マスクは、create_block_causal_mask_optimized 関数によって、Step 1で生成されたsequence_ids に基づいて動的に生成されます。
def create_block_causal_mask_optimized(sequence_ids: torch.Tensor) -> BlockMask:
# sequence_idsは各トークンが属する配列ID
def document_mask(b, h, q_idx, kv_idx): # b: バッチ, h: ヘッド, q_idx: クエリのインデックス, kv_idx: キー/バリューのインデックス
return (
(sequence_ids[b, q_idx] >= sequence_ids[b, kv_idx]) # クエリの配列IDがキー/バリューの配列ID以上であること
& (sequence_ids[b, q_idx] != -1)
& (sequence_ids[b, kv_idx] != -1)
)
# ... (create_block_maskを呼び出してBlockMaskオブジェクトを生成)この論理式 (sequence_ids[b, q_idx] >= sequence_ids[b, kv_idx]) は、クエリ配列(最終シーケンス)が先行する相同配列の参照は許可し、未来の配列情報を参照することは厳密に禁止する(因果性)という、「検索拡張」の要件を保証します。
2.2 Profluent-E1の核心②:カリキュラム学習
Profluent-E1がSOTAを達成した理由の一端は、「カリキュラム学習」にもあると考えられます。
カリキュラム学習:モデルに与える学習データの難易度を徐々に上げていく手法です 。
学習初期: 同時に入力する相同配列の数(K)を少なく(K=2など)、合計トークン長も短く(8192)設定します 。
学習後期: 訓練が進むにつれ、Kを徐々に増やし、最終的には最大K=512、合計長32768という、非常に長く複雑な文脈で学習させます 。
K=2の学習フェーズでは、モデルは文脈情報にほとんど頼れません。そのため、実質的に「単一配列モデル」として機能することを強制され、パラメータに知識を蓄える能力(=単一配列モードの性能)が上がります 。
K=512の学習フェーズでは、モデルは豊富な文脈からその場で推論する能力を獲得し、動的に推論を特殊化する能力(=RAモードの性能)を獲得します 。
src/E1/batch_preparer.py と src/E1/modeling.py が、このような可変長かつ可変配列数(K=2〜512)のバッチを柔軟に処理できる設計になっています。
2.3 リザルト:最高性能(SOTA)の達成
上述のアーキテクチャと学習戦略の有効性が、以下によって示されています。
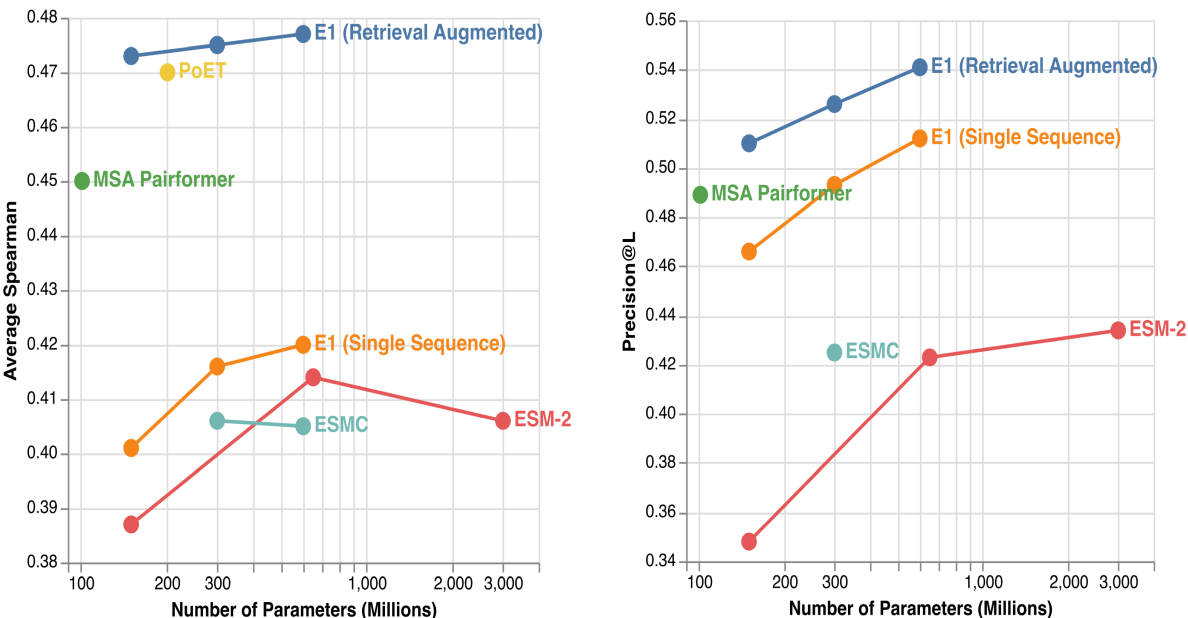
・ゼロショット適応度予測 (ProteinGym): アライメントフリーでありながら、MSAを使用するモデル(MSA Pairformer3)や他のRAモデル(PoET4)を上回り、SOTA(Spearman相関 0.477)を達成しました 。
・教師なしコンタクトマップ予測 (CAMEO): 構造情報を明示的に学習していないにも関わらず、構造特化モデルを超える精度を記録しました 。
パラメータ数はESM-2と同程度〜より小さいモデルであるにも関わらず、高い性能を発揮しています!
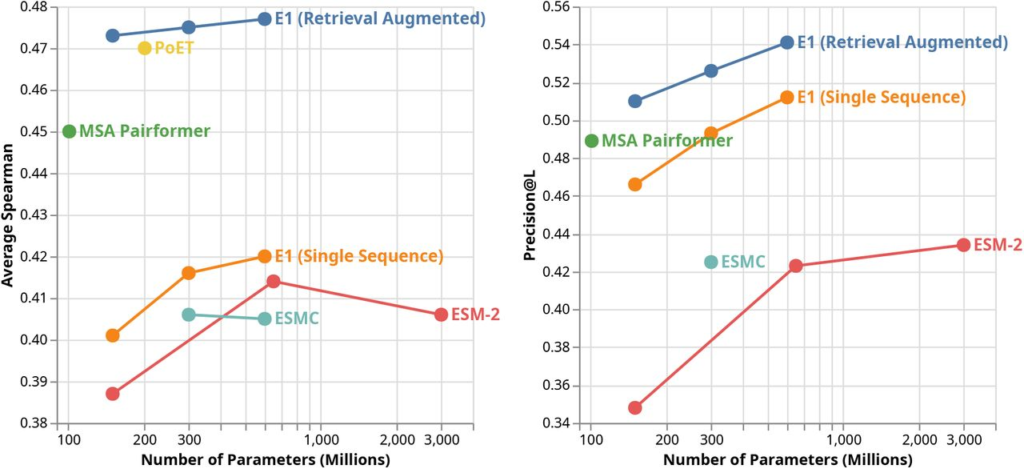
Fig. 2 ProteinGym & CAMEO Performance
引用 Profluent-E1 paper
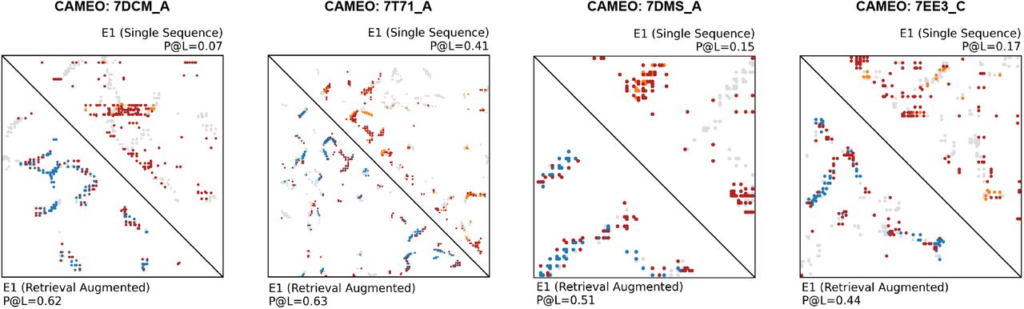
Fig. 3 Contact Map Comparison: RAモードでの劇的な精度向上
引用 Profluent-E1 paper
3. 利用方法の概要
Profluent-E1リポジトリのモデルは、主に以下の2つの方法で利用できます。
コマンドラインツール: src/E1/tools/predict.py や src/E1/tools/score.py を使用して、FASTAファイル形式のタンパク質配列に対して直接予測やスコアリングを実行できます。
Pythonコードからの利用: Hugging Face Transformersライブラリと互換性があるため、Pythonスクリプト内でモデルをロードし、カスタムのデータやタスクに対して柔軟に利用・拡張することが可能です。
学習済みモデルはHuggingFaceからダウンロードして利用でき、150M、300M、600Mというパラメータ数(1.5〜6億)の3つが提供されています。タンパク質言語モデルとしては、使いやすいサイズが実現されています。
また、リポジトリには、タンパク質配列の埋め込み表現を得る方法や、置換変異体の適応度予測の実行例がGoogle Colaboratoryノートブックとして提供されています。
4. まとめと将来展望:マルチモーダル化
Profluent-E1は、アライメントフリーな検索拡張アーキテクチャとその実装により、PLMの新たな基準となる可能性があります。今後はProfluent-E1のマルチモーダル化や、タンパク質生成AIとの共同作業など、様々な応用が期待されます。
また、タンパク質言語モデルの分野におけるカリキュラム学習の適用は、まだ発展途上と考えられます。「シーケンス→構造→機能・局在」といった、モダリティ間の複雑性や情報の階層性に基づく、より高度な「マルチモーダルカリキュラム学習」へと発展する可能性があるかもしれません。
参考文献
1. Lin Z., et al. (2023) Evolutionary-scale prediction of atomic-level protein structure with a language model. Science, 379(6637):1123–1130, doi: 10.1126/science.ade
2. Elnaggar A., et al. (2022) ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans Pattern Anal Mach Intell. doi: 10.1109/TPAMI.2021.3095381
3. Rao, R.M., et al. (2021) MSA Transformer. Proceedings of the 38th In International Conference on Machine Learning, 8844–8856. https://proceedings.mlr.press/v139/rao21a.html
4. Truong T.F. Jr and Bepler T. (2023) Poet: A generative model of protein families as sequences-of-sequences. Advances in Neural Information Processing Systems, 36, 77379–77415, https://proceedings.neurips.cc/paper_files/paper/2023/hash/f4366126eba252699b280e8f93c0ab2f-Abstract-Conference.html

コメント