
1. 研究の背景と目的
タンパク質の構造と機能を自在に設計する技術は、創薬・バイオテクノロジー等の幅広い分野で大きな貢献が期待されています。特に、特定のターゲット分子に結合するタンパク質(バインダー)をゼロから設計するde novoタンパク質バインダー設計は、ターゲットを標的とした治療薬開発の端緒として期待されています。
近年、AlphaFoldに代表される高精度なタンパク質立体構造予測モデルの登場は、配列と構造の関係性の理解を深め、それをタンパク質の設計・デザインに応用する研究が加速しています。例えば、新規タンパク質バインダー設計では、RFdiffusion1、ColabDesign2など多くの手法が開発されています。
RFdiffusion(RoseTTAFold diffusion)は、拡散モデル※による構造生成(A)と逆フォールディングモデルによる配列設計(B)により、タンパク質の設計を行う手法で、タンパク質設計でノーベル賞を受賞されたデイヴィッド・ベイカー先生の研究室で開発されたものです。
※拡散モデル:一般的に画像や音声のような連続値データに対して用いられている生成モデルです。ランダムなノイズを加えたサンプル(拡散過程)から、ノイズ除去(逆拡散過程)を行いながら学習させることで高品質なサンプルを生成します。
A. 拡散モデル(RFdiffusion):連続的な構造生成
・ 主鎖座標の生成: 拡散モデルを用いて、新規なタンパク質の主鎖座標(連続的な3次元空間の情報として)、N, Cα, C, O原子の骨格部分を生成します。
・構造的制約の適用: この主鎖生成プロセスでは、3次元空間における原子の位置(連続値)を扱うことでターゲットタンパク質の表面形状や結合距離などの構造的制約を条件として与えながら、多様な主鎖の可能性を探ることが出来ます。
B. 逆フォールディングモデル(ProteinMPNN3):離散的な配列決定
ProteinMPNN(または類似の逆フォールディングモデル)は、固定された構造(主鎖)が与えられたときに、その構造に折りたたまれると予測されるアミノ酸配列(アミノ酸の種類=離散値)を探索するために最適化されたモデルです。
RFdiffusionは、主鎖の多様な可能性を探ることに長け、複雑なトポロジーでの成功実績もあります。生成した構造(連続値の出力)を、ProteinMPNN(離散値の出力)に渡すという、いわば分業が上手くいく場合も多いです。しかし、ターゲットに対して、バインダーの主鎖構造がその側鎖構造を考慮せずに生成されるため、ProteinMPNNによるアミノ酸配列探索において側鎖を収容できない、「設計不可能な構造」を生成するリスクが高いという課題があります。
Hannes Starkらによって発表されたBoltzGenは、この課題を解決する新たな手法です。BoltzGenは、RFdiffusionが抱えていた連続値による構造生成と離散値による配列決定を組み合わせたことによるリスク、構造決定と配列設計の2段階プロセスによるリスクを排除し、設計と構造予測を単一の全原子生成モデルに統合し、ナノボディ、ペプチド、ミニタンパク質など多様なバインダーを、タンパク質、ペプチド、低分子、核酸といった広範なターゲットに対して設計可能という、ユニバーサル性を特徴としています。
BoltzGen paper: Stark, H. et al. BoltzGen:Toward Universal Binder Design (2025)
chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://hannes-stark.com/assets/boltzgen.pdf
BoltzGen Github: Boltzgen https://github.com/HannesStark/boltzgen
この記事では、BoltzGenの論文と公開されているコードの内容をみていきます。
2. BoltzGenのメソッド、リザルト、および関連コード
2.1. BoltzGenの核心:設計と構造予測の統合、全原子アプローチ
BoltzGenは、例えばRFdiffusionとProteinMPNNで行われる「主鎖生成 → 配列設計」のような2段階プロセスではなく、「設計と構造予測を単一プロセスに統合」した手法となっています。これは、単一のモデルを構造予測と設計(Design)の両タスクで同時に学習させることで実現されています。これによりBoltzGenは、設計プロセス自体がターゲットとバインダー間の相互作用に関する構造的推論能力を持ったモデルとなっています。
この統合を技術的に可能にしたのが、残基タイプの幾何学的エンコーディングという手法です。BoltzGenはアミノ酸の種類を文字列のような離散的なラベルではなく、3次元座標(幾何学)の連続空間で表現します。設計対象の各残基を14個の原子(一部は仮想原子)で表現し、モデルは仮想原子を主鎖原子に対して特定の幾何学的(空間的な)配置に置くことでアミノ酸の種類を指定します(Fig.7)。
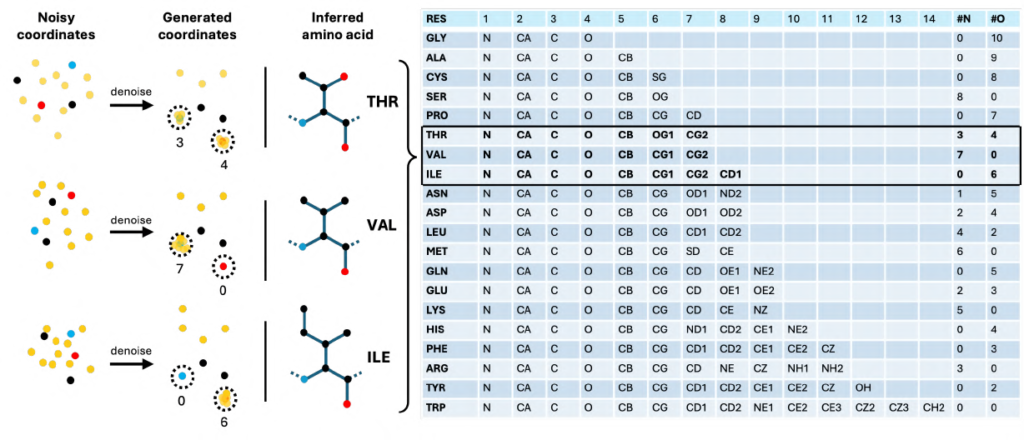
Figure 7: BoltzGenにおける設計残基の14原子による幾何学的エンコーディング
出典 BoltzGen paper
■幾何学的エンコーディングの方法
1. 幾何学的配置とアミノ酸タイプを対応づけるマップ(定数)
設計対象の残基にFig.7に示されている14原子表現が用いられます。側鎖に相当する仮想原子が、主鎖原子(N、Cα、C、O)に重ねて配置された個数のパターンによって、アミノ酸タイプを識別します。
このデコードの対応関係は、const.py 内の辞書に定義されています。
配置パターンの例:
| 残基名 | 参照原子 | 仮想原子マーカーの配置例 | 期待されるカウント (N, Cα, C, O) |
| GLY | N, CA, C, O | [., ., ., ., O, O, O, O, O, O, O, O, O, O] | (0, 0, 0, 10) |
| SER | N, CA, C, O, CB, OG | [., ., ., ., ., ., N, N, N, N, N, N, N, N] | (8, 0, 0, 0) |
この「主鎖原子に重ねて配置された原子の個数」のパターンをキーとし、対応するアミノ酸トークン名(例: SER)に対応させるマップがplacement_count_to_tokenです。
2. 座標からアミノ酸タイプを推論する関数
このデコードを実行するPyTorchベースの関数が、featurizer.py 内のres_from_atom14に実装されています。この関数は、設計されたタンパク質残基の座標を受け取り、以下の処理を実行します。
A. 距離の計算と閾値処理
関数は、設計残基の14原子座標(design_coords)から、主鎖原子(最初の4原子: N, Cα, C, O)と、側鎖/仮想原子(残りの10原子)を切り分けます。
そして、側鎖/仮想原子が、4つの主鎖原子のうちどれに最も近いかを判定するため、原子間の距離を計算します。
# 主鎖原子 (4原子) と側鎖/仮想原子 (10原子) 間の距離を計算
distances = torch.cdist(design_coords[:, :4], design_coords[:, 4:])
# 側鎖原子ごとに最も近い主鎖原子を特定し、その距離 (value) とインデックス (argmin) を取得
value, argmin = torch.min(distances, dim=1)
# 距離が閾値 (threshold) を超える場合(主鎖に重ねて配置されていない場合)、-1とする
argmin[value > threshold] = -1この閾値処理によって、主鎖に重ねて配置された仮想原子のみが選別されます。
B. カウントパターンの生成とデコード
選別された結果に基づき、各主鎖原子(N, Cα, C, O)に重ねて配置された仮想原子の数を集計します(counts)。
# 主鎖原子のインデックス (0, 1, 2, 3) と比較し、マーカーの総数をカウントする。
counts = (argmin[:, :, None] == arange[None, None, :]).sum(1).long() 最後に、この集計されたカウントパターン(例: (8, 0, 0, 0))を、定数マップconst.placement_count_to_tokenを用いて検索し、対応するアミノ酸タイプ(res_type_letters)を決定します。
# カウントパターンを基にアミノ酸タイプを検索(存在しないパターンはUNK)
res_type_letters = [
const.placement_count_to_token.get(tuple(count.tolist()), invalid_token)
for count in counts ] これにより、3次元座標(幾何学)の連続空間に対応した離散的なアミノ酸の種類が表現され、特徴量として更新されます。
2.2. アーキテクチャ、制御性、学習、生成
BoltzGenのアーキテクチャはBoltz-24モデルを基盤とし、AlphaFold35と類似の要素を持ちます。主要な構成要素は、入力情報を処理しトークン/ペア表現を生成するTrunkモジュールと、ノイズが付加された3次元原子座標を反復的にノイズ除去して最終構造を生成するTransformerベースのDiffusionモジュールです。
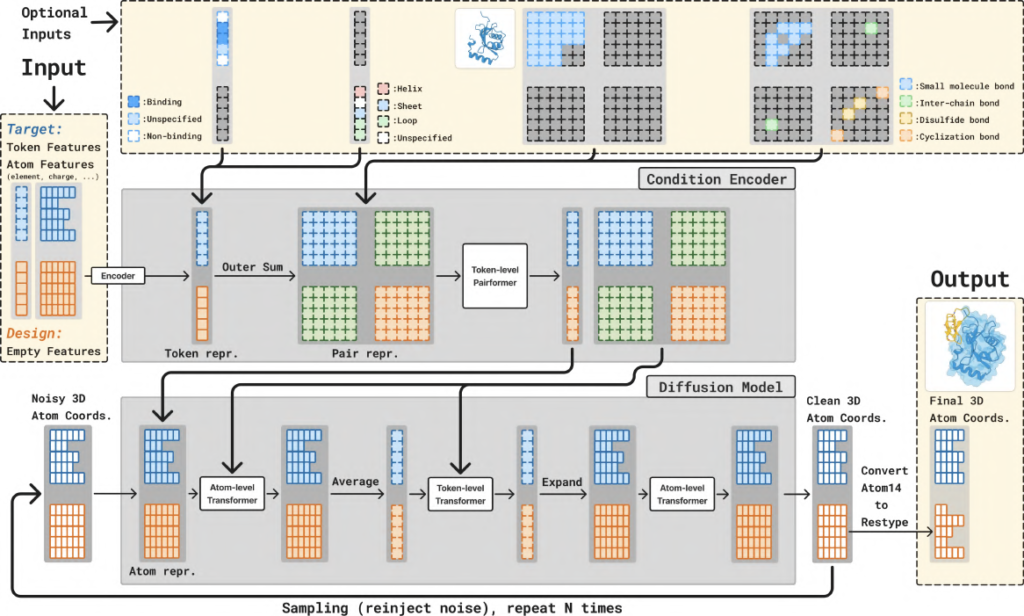
Fig.8: BoltzGenのアーキテクチャ(Condition Encoder(Trunk)とDiffusion Modelの二部構成)
出典 BoltzGen paper
学習によりデノイザー Dθ を獲得し、その Dθ を利用して原子座標 Xの生成を行います。
フェーズ 1: 学習(デノイザーDθ の獲得)
学習の目的は、ノイズ付き座標 Xt から元の原子座標 X0 を推定するネットワーク Fθ(self.score_model)を訓練し、その機能全体としてノイズ除去器(デノイザー) Dθ を獲得することです。 デノイザーDθ は、生成フェーズで使用されます。
1. ノイズの付加と入力の調整 (AtomDiffusion.forward内)
学習用のノイズ σ は対数正規分布からサンプリングされ、これを真の原子座標 atom_coordsに加えます。ネットワークへの入力 rnoisy は、このノイズレベル σ を使って正規化されます (c_in)。
# ノイズ付き座標の生成
noise = torch.randn_like(atom_coords)
noised_atom_coords = atom_coords + padded_sigmas * noise
# ネットワーク入力への事前条件付け (c_in)
net_out = self.score_model(
r_noisy=self.c_in(padded_sigma) * noised_atom_coords,
# ... ) 2. デノイズ座標X̂の推定
ネットワーク Fθ の出力 net_out[“r_update”] は、再度 σ に基づいてスケーリング・結合され、デノイズされた座標 X̂ (denoised_coords) が推定されます。
# デノイズ座標の計算 (c_skip, c_out を使用)
denoised_coords = (
self.c_skip(padded_sigma) * noised_atom_coords
+ self.c_out(padded_sigma) * net_out["r_update"]
) 3. 損失の計算
デノイザー Dθ のパラメータ θ を最適化するため、推定されたデノイズ座標 X̂ と真の座標 X0 (atom_coords_aligned_ground_truth) の間の加重二乗誤差(MSE loss)を計算し、これを最小化します。この損失は、ノイズレベル σ の関数 w(t) で重み付けされます。
# 加重MSEの計算(幾何学的エンコーディングの仮想原子や核酸、リガンドに重みが適用される)
mse_loss = (
(denoised_atom_coords - atom_coords_aligned_ground_truth) ** 2
).sum(dim=-1)
# ... 損失の総和と正規化 ...
loss_weights = self.loss_weight(sigmas)
mse_loss = (mse_loss * loss_weights).mean()この損失関数を最小化することで、ネットワーク Fθ の重み θ が調整され、事前条件付け関数と一体となったノイズ除去器 Dθ の機能が獲得されます。src/boltzgen/model/modules/diffusion.py
フェーズ 2: 生成(EDM Sampler AtomDiffusion.sampleの実行)
生成フェーズでは、学習されたデノイザー Dθ を利用し、ランダムノイズから最終的な原子座標を生成します。
1. サンプリングスケジュールの決定
ノイズレベルを徐々に下げていくスケジュール (sigmas) を設定します。BoltzGenでは、アミノ酸タイプが決定する領域(τi∈[0.6,0.8])での計算を増やすために拡張スケジュール(dilated schedule)が使用されることがあります。
if self.sampling_schedule == "af3":
sigmas = self.sample_schedule_af3(num_sampling_steps)
elif self.sampling_schedule == "dilated":
sigmas = self.sample_schedule_dilated(num_sampling_steps)
# ...2. ノイズの付加とODE方向へのステップ
サンプラーは、各ステップで現在の座標 atom_coords にランダムノイズ eps を加え X̂t (atom_coords_noisy) を得た後、学習済みのデノイザー Dθ(self.preconditioned_network_forward)を使用してノイズ除去予測 X̂ を取得します。
その後、確率流ODEに基づき、デノイズ方向 denoised_over_sigma へ座標を更新し、次のステップX̂t (atom_coords_next) を計算します。
# ステップ 1: ノイズを加える (t_hat = sigma_tm * (1 + gamma) )
eps = noise_scale * sqrt(noise_var) * torch.randn(shape, device=self.device)
atom_coords_noisy = atom_coords + eps
# ステップ 2: モデルによるデノイズ推定を取得
atom_coords_denoised, net_out = self.preconditioned_network_forward(atom_coords_noisy, t_hat, training=False, ...)
# ステップ 3: ODEの方向へ更新し、次の座標を決定
denoised_over_sigma = (atom_coords_noisy - atom_coords_denoised) / t_hat
atom_coords_next = (
atom_coords_noisy + step_scale * (sigma_t - t_hat) * denoised_over_sigma
)
atom_coords = atom_coords_nextこのステップをノイズレベルがゼロになるまで繰り返すことで、最終的な原子座標 (atom_coords)として生成構造を得ます。
フェーズ3として、幾何学からのアミノ酸タイプ推論(デコーディング)(前述、幾何学的エンコーディングの方法参照)が行われます。
2.3. BoltzGenパイプライン
上述したBoltzGenの学習済みモデルが公開され、誰もがバインダー設計を実行できるようになっています。さらに、単なる生成モデルではなく、実験検証に適した最終候補を選抜するための一連のパイプラインとして提供されています。
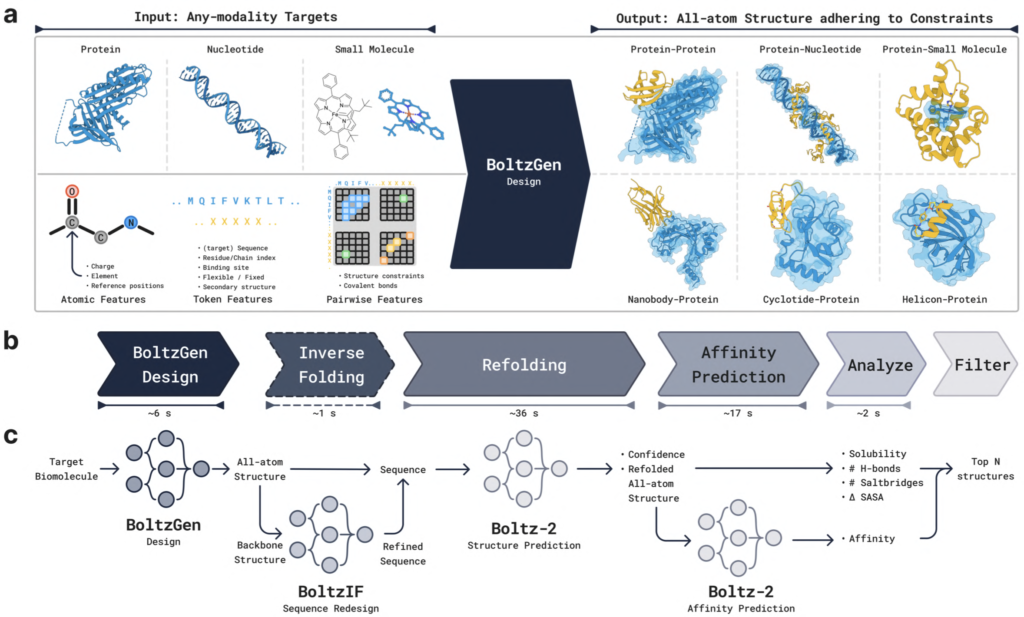
Figure 6: BoltzGenパイプラインの概要
出典 BoltzGen paper
主要なステップは以下の通りです。
1. Design (GPU): BoltzGenの拡散モデル(アーキテクチャ本体)で、初期候補デザインを大量に生成。
2. Inverse Folding (GPU, オプション): BoltzIFモデル(SolubleMPNN類似)で配列を再設計し、溶解性などを改善。
3. Folding (GPU): Boltz-2モデルで設計複合体を再フォールディングし、設計構造とのRMSDを計算。設計の実現可能性を評価する重要なフィルタ。
4. Design Folding (GPU): Boltz-2モデルで設計した配列単独を再フォールディング
5. Affinity Prediction (GPU, 低分子標的時のみ実行): Boltz-2で結合親和性を予測。
6. Analyze (CPU): 水素結合、塩橋、ΔSASA(Delta Solvent Accessible Surface Area)などの物理化学ベースの相互作用スコアや、凝集リスクが高いデザインを排除するための疎水性パッチ面積などが計算される。
7. Filter (CPU): 計算された指標に基づき、Worst weighted metric rank(最悪加重メトリックランク)でデザインをランキングし、品質-多様性選択アルゴリズムで最終候補セットを選抜。
これによって、デザインの生成から厳格な評価と多角的なフィルタリングまでが一貫して実行され、実験的検証に足る、最適化されたデザイン候補群が選抜されます。3、4のステップではBoltz-2が使われます。結合親和性推定が可能なBoltz-2については、下記で詳細を学んでいますのでご参考に。
バインダー設計において優れた成功率を報告しているBindCraft6においても、PyRosetta等を用いた選抜パイプラインが提供されており、類似する設計思想だと思われます。
2.4. 実験結果:新規ターゲットに対する高い成功率とユニバーサル性
BoltzGenにより設計されたバインダーは、ウェットラボでの広範な実験的検証が行わています。特に、学習データに類似構造を含まない新規ターゲット(PDB全体で結合構造を持つ配列同一性30%以上のホモログが存在しない)に対する設計能力が高い、と主張されています。9種の新規タンパク質ターゲットに対し、ナノボディと汎用タンパク質の両方で、ターゲットあたり15個以下の候補評価で66%の成功率(nM親和性バインダー取得)を達成しています。得られた解離定数Kd値は一桁nMレベルに達し、高親和性であることを示しています 。
また、BoltzGenは環状ペプチド設計 (論文 Section 2.6)も可能です。RagA:RagC二量体にバインドするジスルフィド結合環状ペプチドが設計されています(14/24が結合、最高Kd 80 μM)。
さらに、ターゲットのディスオーダー(天然変性)領域へのバインダー設計も試みられています。NPM1のディスオーダー領域へのペプチド設計 (論文 Section 2.4)について、以下のような設定ファイルで、ターゲットのCIFファイルパス、設計するペプチドの長さ範囲(40~80残基)、結合/非結合領域の両者を指定可能です。これにより、ディスオーダー領域に選択的に結合するペプチド設計に成功しています(1/5デザインで成功)。
# NPM1のディスオーダー領域へのペプチド設計:
entities:
- protein:
id: G
sequence: 40..80
- file:
path: npm1.cif
include:
- chain:
id: A
binding_types:
- chain:
id: A
binding: 123..240 #ディスオーダー領域を指定
not_binding: 1..122 #構造形成領域を指定し、ここにバインダーを結合させない。
structure_groups:
- group:
visibility: 1
id: A
res_index: 12..118, 243..291
3. BindCraft/ColabDesignとの比較
拡散モデルであるRFdiffusionやBoltzGenとは異なるアプローチとして、AlphaFold2の構造予測器を「反転」させ、高い予測信頼度(例: ipTMスコア)を与える配列を勾配降下法で探索(最適化)する、ColabDesignが開発されています。BindCraftはこのアプローチで高い成功率を報告しています。BindCraftはAlphaFold2の「信頼度」を最大化する最適化問題として設計を扱いますが、BoltzGenの全原子生成アプローチは、より物理的に忠実な探索を可能にする可能性があります。
*BindCraftについては、以前解説記事を書いていますので、ご参照ください。
また、BindCraftを改良したと報告しているBindEnergyCraft7(*実装コードは現時点では未公開)では、ipTMスコアの代わりに物理化学ベースのエネルギー関数に近い損失関数 pTMEnergy が提案されており、両アプローチとも物理化学的指標を取り込んだモデルへと収束しつつあるのかもしれません。
4. インストール
BoltzGenは、GitHubリポジトリを通じて利用可能で、インストールはcondaを用いた仮想環境構築が推奨されています。一定以上のメモリ(VRAM)を有するGPUを積んだコンピュータにインストールする必要があります。
*Hira Laboでは、RTX A5500(24GB)搭載マシンでBoltzGenパイプラインを完走できることを確認しています。パイプラインのステップ1. Design, 3. Folding, 4. Design Foldingに計算時間の大半を要します。
5. まとめと将来展望
BoltzGenは、設計と構造予測の統合、全原子レベルでの同時生成という革新的なアプローチにより、タンパク質デザインの可能性を大きく拡げたかもしれません。特に、天然変性領域のような難易度の高い標的に対する設計成功は、創薬ターゲットの拡大にも貢献する可能性があります。また、コードや学習済みモデルがMITライセンスのもと公開されており、アカデミアに加えて産業界も利用や改良が可能となっています。
現状のBoltzGenでは、特定の長さのバインダーをデザインした際、ユビキチン様構造への偏りが起こること、オフターゲットへの結合を避ける「選択性」を直接最適化できない点が指摘されています。将来的には、学習データの改善や、生成プロセスを誘導する技術の導入による選択性のさらなる向上が期待されるとのことです。
参考文献
1. Watson, J.L., Juergens, D., Bennett, N.R. et al. De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023). doi: 10.1038/s41586-023-06415-8
2. Ovchinnikov, S. et al. ColabDesign. GitHub (2022) https://github.com/sokrypton/ColabDesign
3. J. Dauparas et al. ,Robust deep learning–based protein sequence design using ProteinMPNN.Science378,49-56(2022).doi: 10.1126/science.add2187
4. Passaro, S. et al. Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction. bioRxiv 659707 (2025) doi: 10.1101/2025.06.14.659707
5. Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024). doi: 10.1038/s41586-024-07487-w
6. Pacesa, M et al. BindCraft: one-shot design of functional protein binders. bioRxiv 615802 (2024) doi: 10.1101/2024.09.30.615802
7. Nori, J. et al. BindEnergyCraft: casting protein structure predictors as energy-based models for binder design. arXiv preprint arXiv:2505.21241 (2025). doi: 10.48550/arXiv.2505.21241

コメント