
近年、大規模言語モデル(LLM)を基盤とする、ChatGPTやGeminiなどの対話型AIシステムが急速に発展しています。しかし、化学や製薬の分野の、特に分子構造のような非テキスト情報を扱う場合、LLMの能力を存分に生かし切れていないところがあります。
本記事では、分子構造情報とLLMを組み合わせることで、薬物に関する多様な質問に回答できるAIシステムDrugChatについて、その技術的な背景と実装を紹介します。
※この記事の作成には、文章の校正の一部で生成AIを活用しています。最終的な内容は、全て筆者が確認・編集しています。
1. 論文の背景とリポジトリの関連性
論文: Liang Y., Zhang R., Li Y., et al., 2024 Multi-Modal Large Language Model Enables All-Purpose Prediction of Drug Mechanisms and Properties doi: 10.1101/2024.09.29.615524
GitHub(リポジトリ): DrugChat: Multi-Modal Large Language Model Enables All-Purpose Prediction of Drug Mechanisms and Properties https://github.com/youweiliang/drugchat
DrugChatの開発の背景として、以下のような課題があります。
・構造-活性相関: 薬物の性質は、分子構造と密接に関連しています。一般的な大規模言語モデル(LLM)は、分子のSMILES表現を単なる文字の羅列として解釈するため、化学的・空間的な文脈を理解することができず、分子内の複雑な構造の関係性を正確に捉えることができませんでした。
・タスク特化型モデル: 分子構造を利用した場合、薬物動態、毒性、結合親和性といった特定の予測タスクごとに特化したモデルの開発が行われています。これらを統合しようとすると、学習コストや管理の複雑さ、多様な予測結果の統合などが必要となってしまいます。
・自由形式テキストへの対応: 従来の薬物情報の予測モデルの多くは、予測結果が離散的なカテゴリに限定されます。適応症や作用機序といった複雑で細やかな意味合いを含む薬学的属性を、自由形式テキストで詳細かつ正確に、表現することができませんでした。
上記の課題に対して、分子構造(SMILES形式)、分子画像、テキストという異なる形式の情報を一括で処理し、分子構造に基づいた対話や情報生成を可能にする分子認識型LLM「DrugChat」が開発されました。DrugChatは、予測結果を自由形式のテキストで生成でき、ユーザーが対話を行うことによって、薬剤やその分子に関する理解を深めることができる対話型AIシステムです。
DrugChatのリポジトリでは、モデルアーキテクチャ、学習コード、推論コード、そしてデモンストレーションを含む、論文で提案された手法の具体的な実装が提供されています。
2. リポジトリの概要・背景
DrugChatの主な目的は、ユーザーが化合物のSMILESを入力し、それに関する様々な質問をすることで、システムが対話形式で回答を生成することです。
以下に示すようなアーキテクチャを採用しています。
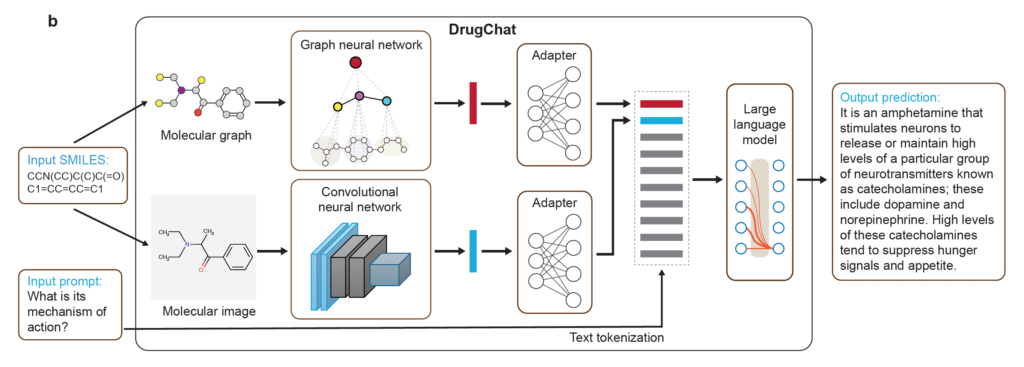
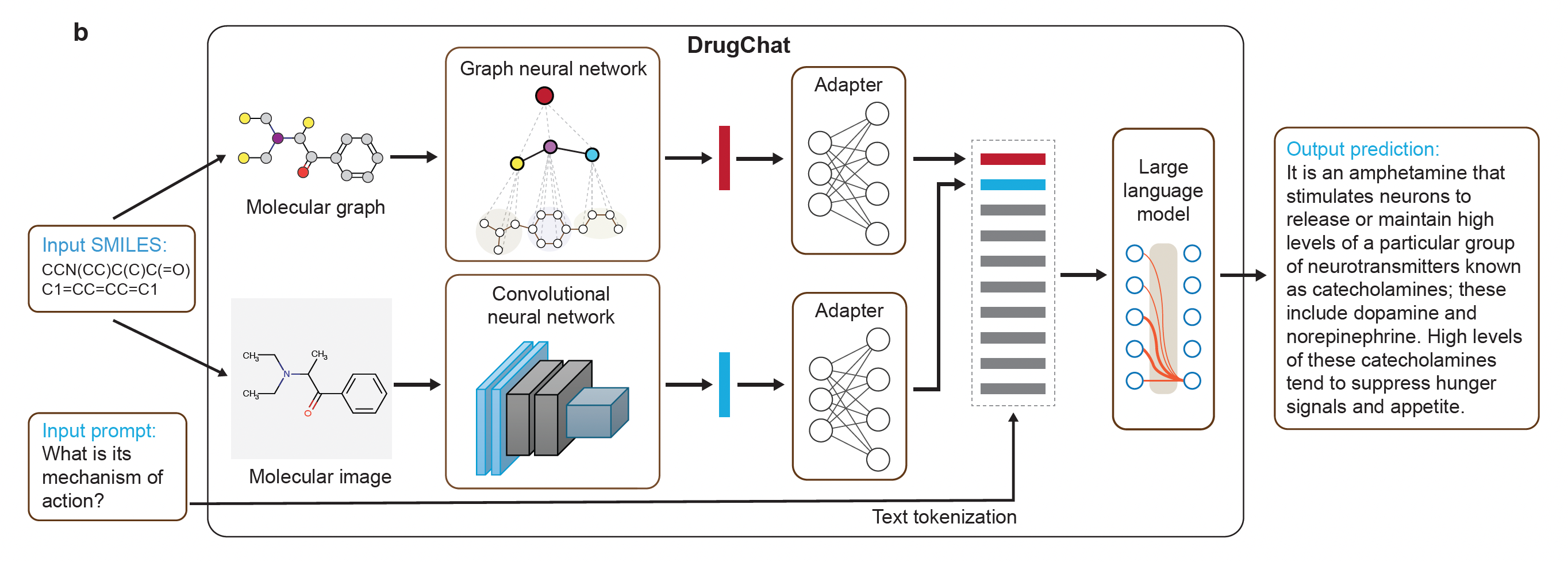
図1:DrugChatのアーキテクチャ概要図
出典: GitHub – youweiliang/drugchat (Copyright 2024 Youwei Liang)
・分子デュアルエンコーダー: 入力されたSMILESは、分子グラフと分子画像の二つの表現に変換されます。分子グラフは自己教師あり学習で事前学習されたグラフニューラルネットワーク(GNN)によって、分子画像は同様に事前学習された畳み込みニューラルネットワーク(CNN)によってそれぞれ表現ベクトル情報に変換(エンコーディング)されます。
・アダプター: これらの分子エンコーディングは、アダプター(多層パーセプトロン)を介してLLMが理解できる(互換性のある)形式、分子トークンと呼ばれる単一ベクトルに変換されます。
・大規模言語モデル (LLM): アダプターからの分子情報とユーザープロンプトがLLMに入力され、最終的な予測(テキスト回答)が生成されます。DrugChatでは、Vicuna-13BをベースとしたLLMが使用されています。
このシステムは、約1.6万個の薬剤分子を含む包括的なデータセットを用いたInstruction tuningによって学習されており、幅広い分子予測タスクに対応可能です。
3. 主要ファイルの概要
DrugChatは、独自のマルチモーダル・分子認識型アーキテクチャと、それを支えるデータ処理・対話管理の仕組みによって機能します。ここでは、論文の主要なMethodsとResultsについて、リポジトリ内の関連ファイルと紐付けながら解説します。
3.1. DrugChatのアーキテクチャと学習プロセス
◆アーキテクチャ:
DrugChatの核となる部分は、分子の化学的・空間的に理解できる分子認識型アーキテクチャであり、以下の要素を統合しています。
・分子表現の抽出:
ー 分子グラフ表現: 分子の接続性や相互作用を捉えるため、50万パラメータを持つ事前学習済みのグラフニューラルネットワーク(GNN)が使用されます。これはZINC15データベースの200万のラベルなし分子を用いた自己教師あり学習により訓練されています。関連ファイルの pipeline/models/gnn.py は、このGNNモデルの定義と実装を含みます。
ー 分子画像表現: 分子の空間パターンや視覚的特徴を捉えるため、1100万パラメータを持つ事前学習済みの畳み込みニューラルネットワーク(CNN)であるImageMol(ResNet-18ベース)が使用されます。これはPubChemデータベースの1000万のラベルなし分子画像を用いた自己教師あり学習により訓練されています。関連ファイルの pipeline/models/image_mol.py は、ImageMolモデルの定義と実装を含みます。
・アダプターによる翻訳: GNNとCNNで抽出された分子表現ベクトルは、それぞれ独立した多層パーセプトロン(MLP)であるアダプターを介してLLMと互換性のある形式に変換されます。アダプターで使われるMLPなどの基本的な部品は pipeline/models/utils.py で定義されています 。
・基盤LLM: Vicuna-13B(Llama-13Bをベースにファインチューニング)が使用されています。関連ファイルの pipeline/models/modeling_llama.py は、LLaMAベースのモデル構造を定義しています。
・DrugChatモデル本体: これら全てのコンポーネントを統合し、分子情報とテキストプロンプトを受け取って応答を生成するモデルの全体像は、pipeline/models/drugchat.py に実装されます。
◆データ収集と学習プロセス:
DrugChatの学習には、ChEMBL、PubChem、DrugBankなどの公開データベースから収集された薬物関連の化合物データが用いられました。91,365組の分子のSMILES文字列、質問、回答の3要素(トリプレット)が学習用に準備されました。RDKitを用いてSMILES文字列から分子グラフを生成し、原子タイプ、キラル性、結合タイプ、結合方向を特徴量として抽出しています。
・SMILESから画像・グラフへの変換:
SMILES文字列からRDKitを用いて分子オブジェクトを生成し、そこから2D画像とグラフ構造(原子をノード、結合をエッジとする)を抽出します。グラフ構造には、原子番号、結合の種類、キラリティなどの詳細な特徴量が付与されます。
関連ファイル:
・dataset/smiles2graph.py: 事前学習や評価用のデータセットとして使うため、SMILESを分子グラフデータに変換・保存します。
・dataset/smiles2image.py: 事前学習や評価用のデータセットとして使うため、SMILESから分子画像を・保存します。
・dataset/smiles2graph_image_demo_realtime.py: 単一のSMILESを分子グラフデータと分子画像に変換します。
・pipeline/datasets/datasets/multimodal_dataset.py: 分子に関する質問応答タスクのために、SMILES、分子画像、分子グラフ、テキストといった複数の種類のデータを統合してAIに供給する分子についてのQAデータセットを定義します。
・pipeline/datasets/__init__.py と pipeline/datasets/datasets/__init__.py: 機械学習において、データを利用するための入り口の役割を担います。
・マルチモーダルデータの前処理とデータ拡張:
生成された画像やテキストデータは、モデルの入力形式に合わせて前処理されます。画像にはリサイズ、クロップ、正規化などの変換が適用され、特に訓練時にはRandAugmentのようなデータ拡張が適用され、モデルの汎化能力を高めます。テキストはトークン化されます。
関連ファイル:
・pipeline/processors/blip_processors.py: BLIPモデル(BLIP2)のための画像とテキストのデータの前処理を定義します。
・pipeline/processors/randaugment.py: 画像のデータオーグメンテーション手法であるRandAugmentの実装を提供し、予測能力を向上させます。
・pipeline/datasets/datasets/base_dataset.py: マルチモーダルデータセットを管理し、前処理を適用する基盤クラスを提供します。
・pipeline/datasets/data_utils.py: 異なるデータセットタイプを効率的に連結・サンプリングするためのユーティリティを提供します。
3.2. 対話システムと共通部品
◆対話システム:
pipeline/conversation/conversation.py と pipeline/conversation/__init__.py は、ユーザーとAIの会話履歴を管理し、対話のロジックを統合する「会話の進行役」として機能します。これらは、会話状態の管理、入力処理と応答生成の連携、対話フローの制御といった主要な機能を提供し、継続的で意味のある対話を可能にする対話型AIシステムの「心臓部」として機能します。
◆共通部品:
システム全体で利用される共通のユーティリティや設定管理は、pipeline/common/ ディレクトリに集約されています。
関連ファイル:
・pipeline/common/registry.py: 様々な部品(モデル、データセットなど)を一元管理し、名前で登録・取得することを可能にします。これによりモジュール性と拡張性を飛躍的に高めています。
・pipeline/common/config.py: システム全体の動作を決める設定(YAMLファイルやコマンドラインからの指示)を読み込み、一元的に管理するための中心モジュールです。
・pipeline/common/logger.py: 学習の進捗や結果を記録します。
・pipeline/common/dist_utils.py: PyTorchを用いた分散学習環境を効率的かつ堅牢に運用するためのユーティリティを提供します。
・pipeline/common/utils.py: データやリソースの管理、およびファイルシステム操作を効率的かつ抽象化された方法で処理するための汎用ユーティリティを提供します。
4. モデルの精度の検証
DrugChatは、薬剤のメカニズムや特性予測において、既存のLLMや他の予測モデルと精度比較をされています。
DrugChatは、薬剤の適応症、薬力学、作用機序、および薬剤概要に関する自由形式のテキスト予測で精度を検証されています。正解との比較スコアとして、薬剤の専門家による評価、意味的類似性、BLEUスコア、METEORスコアなどが算出され、GPT-4と比較して顕著に優れた性能であったことが報告されています。
また、DrugChatは、ヒト細胞への細胞毒性、投与経路(経口、非経口、局所)、プロドラッグの可能性について、「はい」/「いいえ」の離散的カテゴリのスコア(F1スコア)で精度検証を実施されています。GPT-4、LLaMa、ChatGLM、FastChat-T5などのLLM、および分子画像から分子特性を予測するモデルであるImageMolにおいても、F1スコアで大きく上回っています。
このように、薬物属性予測において汎用LLMであるGPT-4を含む他のモデルと比較して大幅に高い精度を実現されています。
5. まとめ
DrugChatは、分子構造という特殊な情報を理解し、それに基づいた対話により、理解を深めることができます。このリポジトリは、そのための先進的なマルチモーダルAIアーキテクチャと、効率的な開発・実行を支えるパイプラインフレームワークを提供しています。
DrugChatは分子認識型LLMであり、薬剤探索だけでなく、将来的には化学反応予測、材料科学など、分子構造の理解が不可欠な様々な分野での応用が期待されます。汎用LLMなどの他のモデルに比べると複数のタスクへの予測精度が高いことから、さらなる予測精度の向上や、多様な予測ツールとの比較や、精度向上が期待されます。
論文では、今後の研究の方向性として、未開拓の薬剤カテゴリを含むより多様な学習データセットの取り込み、分子ドッキングシミュレーションや量子力学計算といった他の計算ツールとの統合による予測能力の強化、そしてLLMによる予測の「解釈可能性」の向上に向けた取り組みが挙げられています。
創薬研究で使われる最先端の予測モデルは、多様な情報を扱う専門的な技術であるため、全てを理解し、使いこなすには非常にハードルが高いです。DrugChatのようなマルチモーダルLLMには、対話を通じて、仕組みを理解しながら最先端のモデルを活用するようなツールになることを期待したいです。例えば、分子情報の入力で、量子力学計算のような高度なツールなどと連携でき、その仕組みや計算についての質問への応答や、計算結果の解釈の提示が可能であれば、研究者だけでなく、薬剤開発に関わる、あるいは学ぶ、幅広い人々への理解が広がることも期待できるように思いました。


コメント