
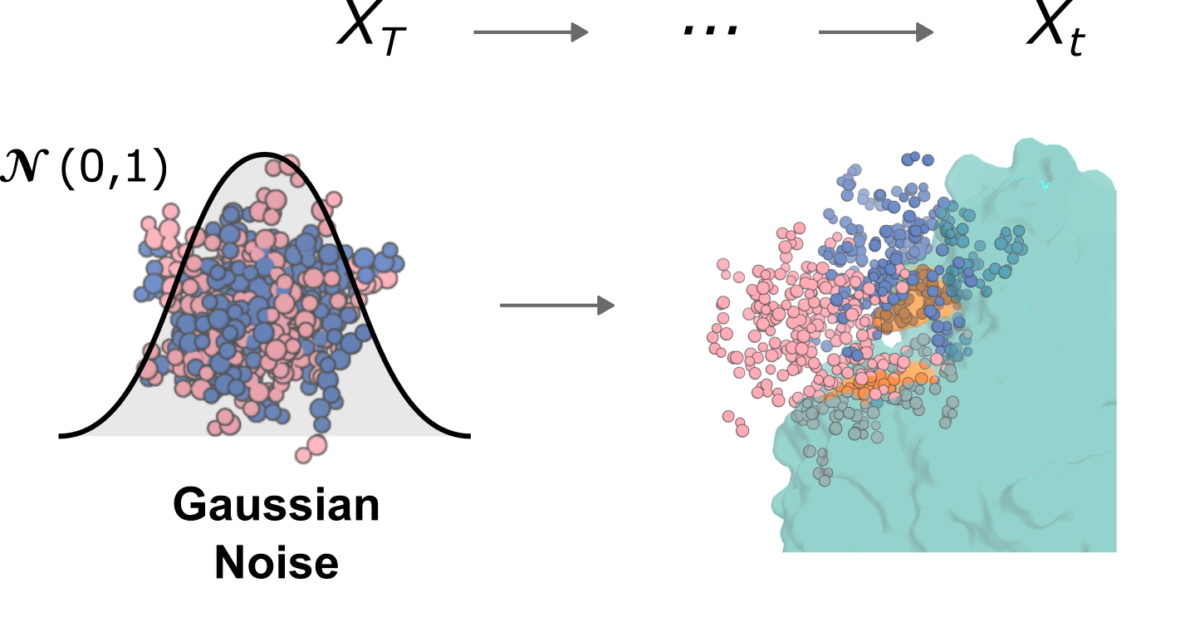
1. 研究の背景と目的
抗体医薬の標的をめぐるジレンマ
免疫系が病原体などを認識する際、抗原の表面にある特定の目印を認識します。この免疫系が認識する最小単位(目印)をエピトープ(抗原決定基)と呼びます 。一方、抗体側がエピトープに結合するために持つ、特異的な結合部位をパラトープと呼びます 。

抗体医薬は、このエピトープとパラトープが高い特異性で結合する抗原抗体反応を応用したものです。がん細胞やウイルスが持つ特定のエピトープだけを精密に狙い撃ちすることで、治療効果を発揮します。
しかし、抗原タンパク質の上には多数のエピトープが存在しますが、そのすべてが薬の標的として有用なわけではありません。「機能的エピトープ」と「免疫優性エピトープ」という考え方があります。
1. 機能的エピトープ(Functional Epitope):薬が狙うべき「真の標的」
抗原の生物学的活性(例:細胞への侵入、受容体への結合、シグナル伝達)に直接的な影響を及ぼす、最も重要な部位を指します。抗体医薬がこの部位に結合すれば、その働きを物理的に阻害し、効果的に治療効果を発揮することが期待できます。
例えば、SARS-CoV-2の場合、ウイルスがヒトの細胞に侵入するために使う受容体結合ドメイン(RBD)という部位が機能的ドメインであり、その中でも特にヒト細胞(ACE2受容体)との結合界面となる部位が、最も重要な機能的エピトープです 。
2. 免疫優性エピトープ(Immunodominant Epitope):免疫系が「応答しやすい標的」
生物学的な重要性(機能)とは無関係に、免疫系が最も強く応答する(=抗体やT細胞を大量に作ってしまう)部位を指します。 抗原表面で最も目立っていたり(例:抗原タンパク質の表面に露出)、B細胞がアクセスしやすい、個人の免疫システム(HLAタイプなど)との相性が良い、といった認識のされやすさを反映します。
抗体設計における最大の壁:「機能」と「優性」の乖離
理想的な抗体医薬は、機能的エピトープを攻撃するものです。しかし、従来の抗体作製法(動物への免疫やファージディスプレイ法など)は、良くも悪くも自然の免疫応答を借用しています。多くの病原体は、この仕組みを逆手に取っています。病原体は、自身の生存に不可欠な機能的エピトープ(例:インフルエンザウイルスの変異しにくいステム領域)を構造的に隠す(=免疫劣性にする)一方で、変異しやすく機能的に重要でない部位(例:インフルエンザの変異しやすいヘッド領域)を意図的に目立たせ、免疫系の攻撃をそこに集中させます。
従来の抗体作製法では、免疫系が応答してしまう「免疫優性エピトープ」に結合する抗体が大量に獲得され、本当に欲しい機能的エピトープ(真の標的)に結合する抗体が獲れないという、根本的なバイアス(偏り)があり、抗体開発における長年の課題でした。
そのため、特定の機能部位を狙い撃ちする「エピトープ特異的」な抗体の、ゼロから(de novo)の設計、計算科学による抗体設計、が期待されてきました。
2025年、この長年の課題に対し、タンパク質生成AIを用いた2つの手法とその成果がCell誌とNature誌に発表されました(1日違い!!)。これらは、AIを活用して「エピトープ特異的な抗体のde novo設計」という同じ目標に挑みつつ、対照的な戦略をとっています。
- MAGE (Cell): ヴァンダービルト大学のグループが開発した「配列ベース」アプローチ。タンパク質言語モデル(PLM)で抗原と抗体の配列間の相互作用の「文法」を学習し、機能的な抗体配列を生成します。
MAGE paper: Wasdin, Perry T., et al. (2025) Generation of antigen-specific paired-chain antibodies using large language models. Cell. doi: 10.1016/j.cell.2025.10.006
MAGE Github: IGlab-VUMC/MAGE_ab_generation https://github.com/IGlab-VUMC/MAGE_ab_generation - RFdiffusion-Ab (Nature): ワシントン大学のノーベル賞受賞者デイヴィッド・ベイカー先生のラボが開発した「構造ベース」アプローチ。抗原エピトープの3D形状に結合するCDR(相補性決定領域)の3D構造を拡散モデルで生成します。
RFdiffusion-Ab paper: Bennett, N.R., et al. (2025) Atomically accurate de novo design of antibodies with RFdiffusion. Nature. doi: 10.1038/s41586-025-09721-5
RFdiffusion-Ab Github: RosettaCommons/RFantibody https://github.com/RosettaCommons/RFantibody
本記事では、これら2つの抗体生成AIについて、その論文と公開リポジトリを詳細に検討しました。
2. 論文とリポジトリ主要ファイルの概要
MAGEとRFdiffusion-Abは、その設計思想、学習データ等において対照的です。
【A】MAGE (Cell): タンパク質言語モデル(PLM)による抗原特異的抗体の生成
MAGE(Monoclonal Antibody Generator)は、「3D構造情報を介さず、膨大な配列データのみから抗原と抗体の関係性をPLMに学習」させています。具体的には、抗原配列(コンテキスト)から、それに結合するペア型抗体(VH/VL)配列を生成するよう、Progen23をファインチューニングしています。
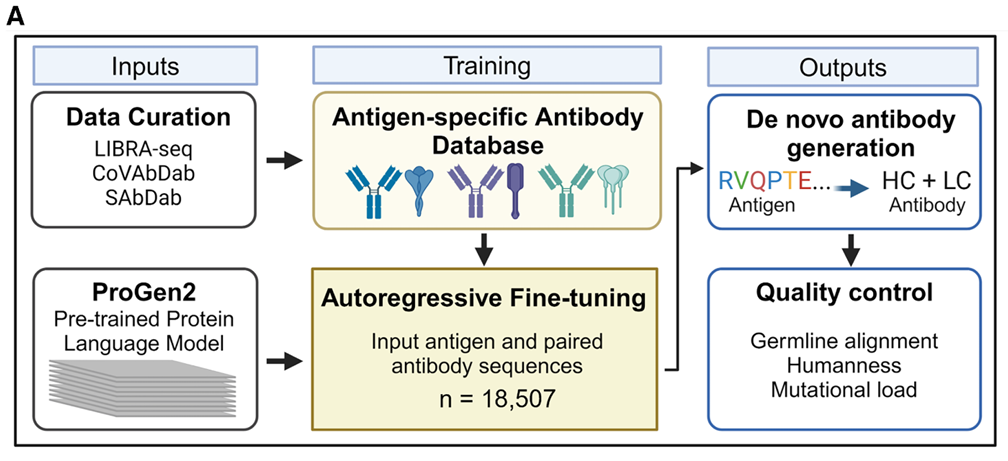
Fig 1:MAGEのワークフロー
引用: MAGE paper
論文の核心とオリジナリティ:
1. 高品質なデータセットの構築
MAGEでは、合計18,507ペアに及ぶ高品質な「抗原配列と抗体配列のペア」のデータが構築されています。このデータセットは、CoV-AbDab、SAbDab、PLAbDabなどの公開データベース からキュレーションされたデータと、独自のデータから構成されています。その独自データは、LIBRA-seq(Linking B cell receptors to antigen-specificity through sequencing)技術を用いて取得された1,924ペアのデータです。LIBRA-seqは、単一のB細胞受容体(BCR、すなわち抗体配列)とそれが結合する抗原特異性を直接マッピングする手法であり、質の高い学習データが追加されています。
2. PLMへの入力形式と学習タスクの定義
Progen2は、抗原配列、重鎖(VH)、軽鎖(VL)を特殊トークンで連結した単一のシーケンスを入力として、ファインチューニングされています。特殊トークン[SEP](セパレーター)と[LC](軽鎖マーカー)をトークナイザーに追加し、訓練データは以下の形式に整形されます。
<|bos|> [抗原配列] [SEP] [重鎖VH配列] [LC] [軽鎖VL配列] <|eos|>
これにより、モデルは「与えられた抗原配列の文脈に続いて、適切な抗体配列(VHとVL)を生成」するタスクを学習します。Progen2-baseは、この連結シーケンスを処理するのに十分な2,048トークンのコンテキストウィンドウを持っています。
3. 損失計算のカスタマイズ:masked_sep_lossの実装(Fine_tuning/full_model_training_24-03-12.py)
モデルの学習を抗体配列の生成精度のみに集中させるために、損失関数がカスタマイズされています。抗原配列部分([SEP]トークンより前)の予測損失を意図的にマスク(無視)し、抗体配列(VHおよびVL)の負の対数尤度損失(NLL Loss)のみを計算しています。このロジックは、masked_sep_loss関数として実装されています。
def masked_sep_loss(model, batch, device):
# ... (targets, output logits, sep_token_idを定義)
# SEPトークンの位置を特定
sep_positions = (targets == sep_token_id).long().argmax(dim=1)
# sep_mask: SEP位置より後方をTrueとする
sep_mask = positions > sep_positions.unsqueeze(-1)
pad_mask = targets != pad_token_id # パディングトークンでない場所をTrue
mask = sep_mask & pad_mask # SEPの後方 かつ パディングでない場所に限定
# NLL Lossを計算
loss = F.nll_loss(log_probs.transpose(1, 2), targets, reduction='none')
# マスクを適用し、抗体配列部分の損失のみを残す
masked_loss = loss * mask.float()
# マスクされた損失の平均を最終損失とする
final_loss = masked_loss.sum() / mask.sum()
return final_lossこの処理により、モデルは抗原配列をコンテキストとしてのみ利用し、実際に生成される抗体配列の品質向上に特化して重みが調整されることになります。
*コード上のIssue?
masked_sep_loss関数を定義しているCustomNLLEntropyLossTrainerクラスが、HuggingFace Trainerのデフォルト損失計算を置き換えるためのcompute_lossメソッドをオーバーライドしていないようです。定義されたmasked_sep_loss関数が学習ループで使用されず、デフォルトの損失計算が実行されているかも?
論文リザルト:
MAGEは、配列情報のみから驚くべき成果を上げています。
・高いヒット率: SARS-CoV-2 RBD (ヒット率 9/20, 45%)、RSV-A F (7/23, 30%)、H5/TX/24 HA (5/18, 28%)という3種の病原体に対し、実験的に結合が確認できる抗体を高確率で生成。
・ゼロショット能力: 特にH5/TX/24株は訓練データに含まれておらず、MAGEは関連株の学習データに基づき、この未知の株に対しても機能的な抗体をゼロショットで生成することに成功。
・創発的な構造的多様性: MAGEは3D構造を明示的に学習していませんが、生成された抗体は構造的に多様。RSV-A Fに対して生成された2つの抗体(RSV-2245, RSV-3301)のCryo-EM構造により、これらが全く異なるエピトープ(抗原性サイトVとサイトI)に、異なる様式で結合していることが確認されています。
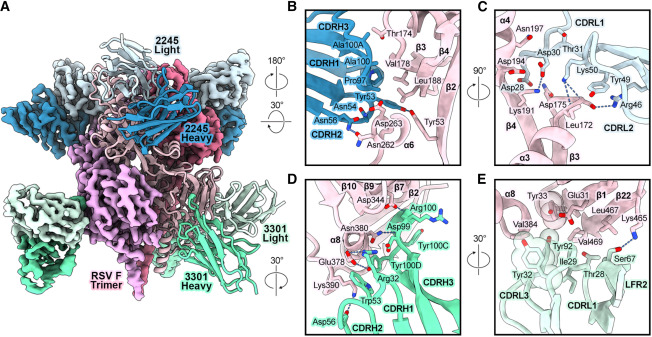
Fig 7 RSV-2245/RSV-3301のCryo-EM構造
引用: MAGE paper
この結果は、MAGEが抗原配列という文脈から、それに結合しうる多様な抗体配列の「確率分布」を学習しており、多様な抗体を生成できる可能性を示しています。
【B】RFdiffusion-Ab (RFantibody, Nature): 原子レベルの精度を持つ de novo 抗体設計
RFdiffusion-Abは、抗原エピトープの3D構造に対し、原子レベルで構造的に適合するCDRの3D形状を直接設計します。
論文の核心とオリジナリティ:
①拡散モデルの抗体特化: タンパク質の主鎖構造生成モデルRFdiffusionを、抗原-抗体複合体の構造データベース(SAbDab等)でファインチューニング。これにより、オリジナルのRFdiffusionでは不可能だった、抗体特有の柔軟なCDRループ、特にCDR H3を用いた界面のde novo設計が可能に。
②エピトープ特異的な構造設計: ユーザーが指定した抗原3D構造上の「ホットスポット」(エピトープ)に対し 、(A) CDRループのde novo構造 と (B) 抗体フレームワーク全体のドッキング(位置・向き)を、拡散プロセスを通じて同時に生成・最適化します。
③検証フィルター(オラクル)の自作: De novo設計の最大の課題は、生成された構造が真の構造なのか検証する「オラクル」の不在です。開発時、AlphaFold2は抗原-抗体複合体の予測精度が低く、使えませんでした。そこでベイカー先生のラボが開発したRoseTTAFold2(RF2)を、抗体複合体データで独自にファインチューニングしたものを用いています。これを「自己一貫性」(設計配列をRF2-finetunedで予測した構造が、元の設計構造と一致するか)を検証する「オラクル」としています。

Fig 1e RFdiffusion-Abのパイプライン図
引用: RFdiffusion-Ab paper
RFdiffusion-Abのワークフローは、以下のStep1~3の3つの異なるAIモデルを連携したパイプラインです。
・Step 1: 構造生成 (scripts/rfdiffusion_inference.py) の実装詳細
構造生成プロセスは、主に「設計ループの管理」「逆拡散ステップの実行」「ホットスポットターゲティングの検証」という3つの重要な要素で構成されます。
1. 設計ループの管理 (Design Loop Management)
スクリプトのメイン関数では、設定ファイルで指定された数(num_designs)だけ抗体を生成するための外部ループが実行されます。これにより、複数の候補構造が系統的にサンプリングされます。
for i_des in range(design_startnum, design_startnum + sampler.inf_conf.num_designs):
# ... (デザイン開始時間の記録など)
out_prefix = f'{sampler.inf_conf.output_prefix}_{i_des}'
log.info(f'Making design {out_prefix}')
# ... (cautious modeでのスキップ処理)
failed=0
while True:
x_init, seq_init = sampler.sample_init()
# ... (中間結果のスタック初期化)
# ...
# Loop over number of reverse diffusion time steps.
for t in range(int(sampler.t_step_input), sampler.inf_conf.final_step-1, -1):
# ... (拡散ステップの実行)
# ... (ホットスポットターゲティングの検証と、失敗時のループ継続)
# break out of while loop
breakこの構造は、設計番号(i_des)に基づいて個々のデザインを生成する外側のループ と、構造が設計条件を満たさない場合に再試行を可能にする内側の while True ループ から成ります。
内側のループでは、まず sampler.sample_init() を呼び出して、設計対象領域(CDR)にノイズが付加された初期座標 (x_ini>) と初期配列 (seq_init) が生成されます。このノイズ状態 (xT) から逆拡散プロセスが開始されます。
2. 逆拡散ステップの反復実行 (Iterative Denoising)
構造生成の核心は、時間ステップ t に従ってノイズを段階的に除去し(逆拡散)、ノイズの多い状態 (x_t) から洗練された構造 (px0) へと収束させるプロセスです。
x_t = torch.clone(x_init)
seq_t = torch.clone(seq_init)
# Loop over number of reverse diffusion time steps.
for t in range(int(sampler.t_step_input), sampler.inf_conf.final_step-1, -1):
px0, x_t, seq_t, tors_t, plddt = sampler.sample_step(
t=t, seq_t=seq_t, x_t=x_t, seq_init=seq_init, final_step=sampler.inf_conf.final_step)
px0_xyz_stack.append(px0)
denoised_xyz_stack.append(x_t)
# ...この for t in range(…) ループが、逆拡散プロセスを実施します。
・ ループは最大ノイズステップ (sampler.t_step_input) から最終ステップ (sampler.inf_conf.final_step) まで、時間を巻き戻す形で実行されます。
・ 各ステップで sampler.sample_step が呼び出されます。このメソッドは、現在のノイズレベル (t)、座標 (x_t)、配列 (seq_t) を入力として受け取り、モデルの予測(ノイズ除去された構造 px0)と、次に低いノイズレベルでの新しい座標 (x_t) および配列 (seq_t) を返します。
・ これにより、CDRバックボーンの剛体フレームとドッキング角度が、ステップごとに幾何学的な等変性を保ちながら洗練され、最終的な構造へと導かれます。
3. ホットスポットターゲティングの検証 (Hotspot Targeting Validation)
RFdiffusion-Abの設計目標は、単に構造を生成することではなく、ユーザーが指定したエピトープ(ホットスポット)をターゲットとする構造を生成することです。そのため、設計の途中で、生成されたCDRループがホットスポットに物理的に接近しているか検証されます。
# Cb = generate_Cbeta(N=px0[:,0], Ca=px0[:,1], C=px0[:,2]) # Cβ原子座標の生成
# ...
dist = torch.cdist(Cb[sampler.ab_item.hotspots], Cb[sampler.ab_item.loop_mask]) # [hotspot_L, loop_L]
mindist = torch.min(dist, dim=1).values # The min distance for each hotspot
overallmin = torch.min(mindist) # The distance of the closest hotspot to a loop
print(f'Overall min distance hotspot to designed loop: {overallmin}')
if conf.antibody.terminate_bad_targeting == t and overallmin > conf.antibody.hotspot_termination_threshold:
print("Not targeting correctly")
failed+=1
if failed>=conf.antibody.hotspot_termination_failures_permitted:
# ... (システム終了)
continue ①まず、生成された主鎖原子 (N, Ca, C) からCβ原子 (Cb) 座標が推定されます。
②torch.cdist を使用して、抗原上のホットスポット残基のCβと、生成されたCDRループ残基のCβとの間の距離行列が計算されます。
③ 最も近いホットスポットとループ残基間の距離 (overallmin) が特定され、この最小距離が、設計が成功しているかどうかの指標となります。
④ もし、特定のタイムステップ (t) において、この最小距離が許容閾値 (hotspot_termination_threshold) を超えている場合、ターゲットを正しく捉えていないと判断され、そのデザインは失敗としてカウントされ、while True ループの先頭に戻って再試行されます。
このように、RFdiffusion-Abは、確率的なノイズ除去と、ホットスポットターゲティングという物理的な制約を組み合わせることで、特定の標的に対して結合する能力を持つ主鎖構造を効率的に生成するように実装されています。
Step 2: 配列設計 (scripts/proteinmpnn_interface_design.py)
・Step 1で生成された「構造(PDB)」を入力とし、ProteinMPNN_runnerがその構造に適合する「配列」を設計します(逆折り畳み)。
・-loop_string引数で設計対象のCDR(例: “H1,H2,H3″)を指定し、フレームワーク配列は固定します 。
Step 3: 検証フィルター (src/rfantibody/rf2/modules/model_runner.py) の実装詳細
1. AbPredictorクラスによる反復的洗練
検証フィルターは、AbPredictorクラスによって実現されます。これはRF2を抗体設計向けに特化させたものです。
AbPredictorは、入力された配列から構造を予測する際に、「リサイクル」と呼ばれる反復的な予測ループを実行し、段階的に構造予測の精度を向上させます。
with torch.no_grad():
for i_cycle in range(self.conf.inference.num_recycles + 1):
# ... input/output handling removed for brevity ...
output_i=self._output_dictionary(self.model(**input_i), input_i)
metrics_i=self._process_output(output_i, output_pose_i, pose)
if metrics_i['pred_lddt'].mean() > best_lddt.mean():
# ... tracking best pose ...このループでは、モデルは各ステップの予測結果を、pLDDT (Predicted Local Distance Difference Test)の平均値に基づいて評価し、最も信頼性の高い構造(best_pose)を追跡します。
2. 設計品質の評価指標(pLDDTとpAE)
設計の信頼度を測る主要な指標は、pLDDTとpAE (Predicted Aligned Error)です。これらは、予測された構造の品質スコアです。
特に、interaction_paeは、抗原と抗体間での平均残基アラインメント誤差を示しており、予測された結合界面の品質を評価する重要な尺度です。
def get_confidence_scores(self, output_i: dict, pose: Pose, metrics: dict) -> None:
# ... pred_lddt calculation removed for brevity ...
pae = pae_unbin(logits_pae)
interaction_pae=pae[0, ~pose.same_chain].mean()
metrics["interaction_pae"] = interaction_pae
metrics["pae"] = pae
metrics["pred_lddt"] = pred_lddt3. 自己一貫性の定量化(RMSD)
検証フィルターは、予測された構造(pose1:設計配列による折り畳み結果)と、元の設計構造(pose2:RFdiffusion-Abで生成されたターゲット構造)との間の構造的類似性(RMSD)を計算します。これにより、設計された配列が意図したコンフォメーションに実際に折り畳まれるかの定量化を試みています。
def get_rmsds(pose1: Pose, pose2: Pose, metrics: dict) -> None:
# target-aligned rmsds removed for brevity
# framework-aligned monomer rmsds
rmsd.tmalign_to_subset(pose1, pose2, subset='framework')
metrics['framework_aligned_antibody_rmsd'] = rmsd.calc_prealigned_rmsd(pose1, pose2, pose1.antibody_mask)
metrics['framework_aligned_cdr_rmsd'] = rmsd.calc_prealigned_rmsd(pose1, pose2, pose1.cdrs.mask_1d)
# individual loop rmsds removed for brevityRMSDが低いほど予測構造は元の設計に近く(高い自己一貫性)、低いRMSDと高い品質(低いpAE)を持つデザインが、最終的な成功候補として選定されます。
論文リザルト:
RFdiffusion-Abは、構造ベース設計の「精度」と「制御性」を実証しています。
・原子レベルの精度: Cryo-EMで決定された実験構造が、計算上の設計モデルと高い精度で一致しました 。
ー VHH_flu_01 (vs インフルエンザHA): 設計が最も困難なCDR3ループのRMSDがわずか0.8Å 。
ー scFv6 (vs TcdB): de novoで設計された6つ全てのCDRループが、原子レベルの精度で設計モデルと一致(例: CDRL1/L3で0.2Å RMSD)。
・ピンポイントなエピトープ制御: ユーザーが指定したエピトープを正確に標的に。
ー TcdBのFrizzledインターフェース(PDBに抗体結合例がなかった部位)。
ー PHOX2Bペプチド-MHC複合体の「R6残基」という特定のアミノ酸。
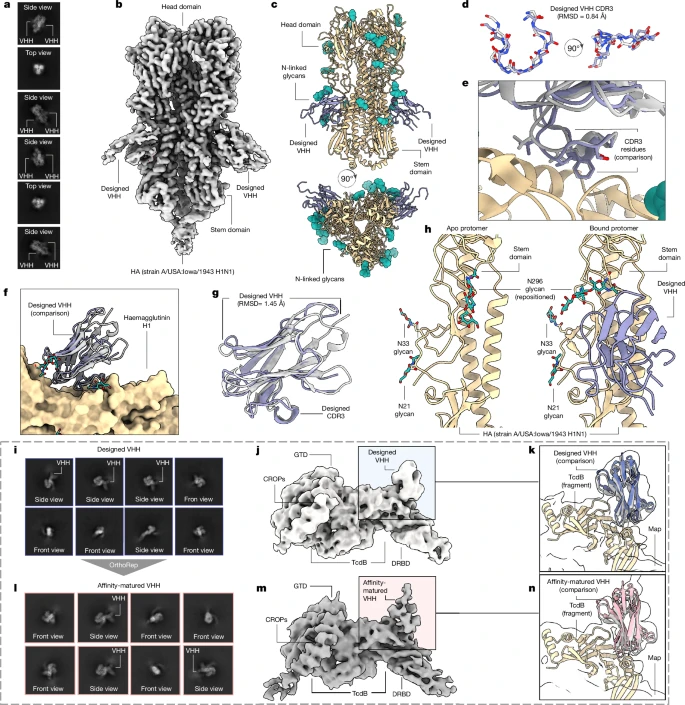
Fig. 3 VHH_flu_01
引用: RFdiffusion-Ab paper
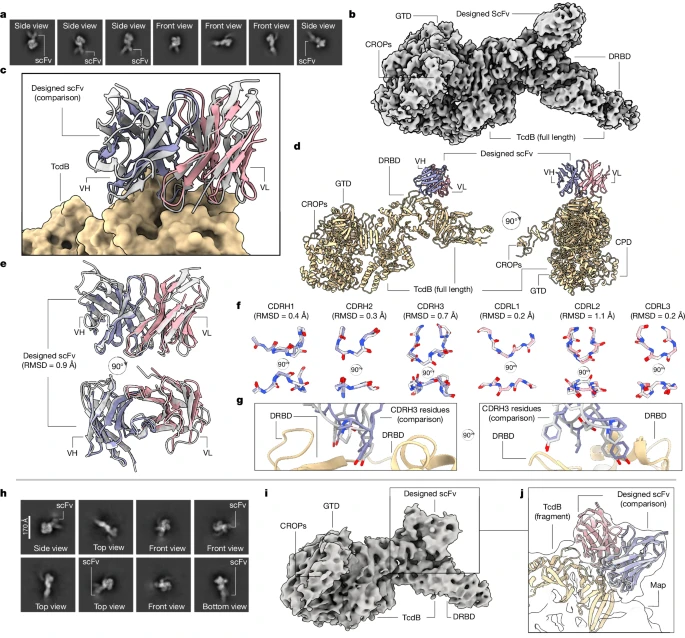
Fig. 5 scFv6 Cryo-EM構造比較図
引用: RFdiffusion-Ab paper
補足:周辺研究から見る「オラクル問題」の現在地
RFdiffusion-Abが直面した「設計検証(オラクル)の困難さ」は、抗体生成AIに関する大きな課題です。論文執筆後(2024年)に発表されたAlphaFold3 (AF3) でもこの問題は解決されていません。
最新の研究によれば、AF3による抗原-抗体複合体予測において、高精度(DockQ ≥ 0.80)予測の成功率は11%に留まっています5。CDR H3ループのモデリング精度が高くない(RMSD中央値 ≈ 2.73Å)ためです。この事実からも、RFdiffusion-Abが独自の検証フィルター(RF2-finetuned)を用意する必要があったことがわかります。
3. ワークフローの対比
| MAGE (Monoclonal Antibody Generator) | RFdiffusion-Ab (RFdiffusion for antibody design) | |
| 基盤技術/設計手法 | 配列ベースの大規模言語モデル | 構造ベースの拡散モデル |
| 入力データ | 標的抗原のアミノ酸配列(文字列) | 標的抗原の3D構造(PDB)・標的としたいエピトープの残基番号リスト |
| 主な出力データ | VH/VLペアの抗体配列(アミノ酸配列の文字列、CSV形式) | 3D構造モデル(PDB形式)および対応するCDR配列 VHH、scFv、完全抗体(IgG)の設計が可能である。 |
| 検証・フィルタリング | 配列検証・ヒトらしさによるフィルタリング 生成された配列をANARCI(配列検証)やBioPhi プラットフォームのOASis(ヒトらしさ)でフィルタリングを行う。 | 検証/フィルタリング③ -設計構造の検証: ファインチューニングされたRF2を使用し、設計された配列が意図した構造と一致するかを評価。 |
4. まとめ と将来展望
MAGEとRFdiffusion-Abは、AIによる抗体de novo設計の2つの異なるパラダイムを確立しました。
しかし、これらの研究は「終わり」ではなく、次世代AIの「始まり」です。
1. オラクル問題: AF3の現状が示す通り、de novo設計された構造の妥当性をin silicoで高速に検証する手段は依然としてボトルネックです。
2. ペア鎖 vs エピトープ制御: MAGEはペア鎖(VH/VL)を生成できますがエピトープを制御できず、RFdiffusion-Abはエピトープを制御できますがペア鎖のde novo設計はVHH(単鎖)より困難でした。
MAGEとRFdiffusion-Abがそれぞれ解いた「問題の半分」を統合する可能性があるのが、次世代のマルチモーダルAIです。例えば、ESM36: EvolutionaryScaleが発表したESM3は、配列・構造・機能の3モダリティを単一のトークンスペースでLLMです。これにより、MAGEができなかった「特定エピトープの構造」や「特定の機能(例:中和活性)」をプロンプトとして条件付けし、抗体配列を生成できる可能性があります。
また、拡散モデルによる抗体生成AIとして、側鎖を含む全原子を生成可能なBoltzGenが公開されています。

今後のさらなる発展が期待されます。
参考文献
1. Abramson, J. et al. (2024) Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500. doi: 10.1038/s41586-024-07487-w
2. Hayes T, et al. (2025) Simulating 500 million years of evolution with a language model. Science. 2025, 387, 850-858. doi: 10.1126/science.ads0018.
3. Nijkamp, E., et al. (2023) ProGen2: Exploring the boundaries of protein language models. Cell Systems, 14, 968 – 978.e3. doi: 10.1016/j.cels.2023.10.002
4. Prihoda D., et al. (2022) BioPhi: A platform for antibody design, humanization, and humanness evaluation based on natural antibody repertoires and deep learning. MAbs. 14, 2020203. doi: 10.1080/19420862.2021.2020203.
5. Hitawala FN, Gray JJ. (2025) What does AlphaFold3 learn about antigen and nanobody docking, and what remains unsolved? bioRxiv [Preprint]. doi: 10.1101/2024.09.21.614257
6. Thomas Hayes et al. ,Simulating 500 million years of evolution with a language model.Science387,850-858(2025).DOI:10.1126/science.ads0018


コメント