
このカテゴリーの記事では、「Pythonを使った機械学習やケモ・バイオインフォマティクスの実装や論文」を紹介していきたいと考えています。Python は3系(3.7)、anacondaを中心にして環境構築していきます。以下のようなハード・ソフト環境(CentOS Linux)を用いますが、Python(anaconda)が利用できれば、異なる環境下でも類似の実装が可能かと思います。
● CPU Intel® Core™ i7-6700K CPU @ 4.00GHz × 8
● メモリ 64GB
● GPU GeForce GTX 1080/PCIe/SSE2
● OS CentOS Linux 8
● Python 3.7
はじめに
前々回、前回の記事で、conda を利用して、BLASTによる相同性検索を実行できる Local な仮想環境を構築し、ヒトの全タンパク質(推定の物も含む)の一次構造をデータベースとして設定しました。さらに、この環境を使って、出芽酵母の全タンパク質やチンパンジーの1番染色体にコードされているタンパク質を問合せ配列(Query(クエリ))としたBLAST検索を行い、結果をざっくりとまとめてみました。

今回、BLAST検索結果の分析として、データ解析用ライブラリ Pandas や 、Python や R の対話型実行環境で、分析手法をブラウザ上で管理できる Jupyter notebook 等を導入したいと思います。これらツールも、これまで通り conda で簡単に利用できます。
BLAST結果のgrepによる処理
Jupyter notebook 等を使って行く前に、まず、BLAST結果から必要な情報を抽出したいと思います。前回の記事では、下記のコマンドで出芽酵母の全タンパク質 6002個をクエリとしたBLAST検索しました。
$ blastp -query GCF_000146045.2_R64_protein.faa -db GCF_000001405.39_GRCh38.p13_protein.faa -out yeast.out -num_threads 8 -evalue 0.001 -num_alignments 1
結果として6002個のうち、約57%に相当する3450個のタンパク質がヒトの何らかのタンパク質にヒットし、E value 0.001以下を示す類似タンパク質がヒトゲノム中に存在する可能性があることがわかりました。この結果は、yeast.out に書き込まれています。また、-num_alignments 1 を指定することで、3450個それぞれと、トップヒットしたヒトのタンパク質とのアライメント結果も保存されています。今回、このアライメント結果を分析してみましょう。まずは、下記のコマンドで、yeast.out から情報を抽出してみましょう。
$ cd /home/xxxx/human_protein ## 前回BLAST検索を実行したディレクトリに移動します。 $ grep -A 10 '>' yeast.out | grep -e '>' -e ' Identities = '
この grep コマンドを利用することで、下記のような表示が得られます。
[xxxx@localhost ~/human_protein]$ grep -A 10 '>' yeast.out | grep -e '>' -e ' Identities = ' >NP_006834.2 L-serine dehydratase/L-threonine deaminase [Homo Identities = 117/328 (36%), Positives = 175/328 (53%), Gaps = 26/328 (8%) >YP_003024028.1 cytochrome c oxidase subunit I (mitochondrion) Identities = 301/529 (57%), Positives = 398/529 (75%), Gaps = 25/529 (5%) >YP_003024028.1 cytochrome c oxidase subunit I (mitochondrion) Identities = 214/324 (66%), Positives = 265/324 (82%), Gaps = 3/324 (1%) ・ ・ ・
> で始まる行がトップヒットしたヒトのタンパク質の名前です。次の行が、アライメントの結果をまとめたもので、Identities, Positives, Gaps について、アミノ酸残基数とその割合が示されています。この2行1組の結果が、タンパク質3450個分表示されています。
さらに grep コマンドを以下のように変更することで、Identities の情報のみを抜き出すことが可能です。
$ grep -A 10 '>' yeast.out | grep -e ' Identities = ' | grep -oP '^.*?\)' | grep -oP '\([0-9]*%\)'
このコマンドで、下記の出力が得られます。
[xxxx@localhost ~/human_protein]$ grep -A 10 '>' yeast.out | grep -e ' Identities = ' | grep -oP '^.*?\)' | grep -oP '\([0-9]*%\)' (36%) (57%) (66%) ・ ・ ・
Identities = XXX/XXX の後ろに表示されていた、アライメントにおける一致の割合(%)の部分を抜き出すことができました。3450個分の%が1列に得られています。
さらに先程のコマンドの最後に、 >> ファイル名 とすれば、端末への出力をテキストファイルとして保存することができます。
$ grep -A 10 '>' yeast.out | grep -e ' Identities = ' | grep -oP '^.*?\)' | grep -oP '\([0-9]*%\)' >> Identities.out
ファイル名は、Identities.out としました。 grep コマンドの使用法を色々と変更することで、 yeast.out から他の情報を抜き出すことも可能です。
また、このブログでは主にPython や R を用いた分析を紹介しますが、このファイルをエクセル等、他の表計算で分析することも可能です。
Pandas, Jupyter notebook, Plotly 等のインストール
次に使用するパッケージ群を準備します。前回までに仮想環境 blast を構築済みであれば、この環境に追加でインストールしてしまいましょう。下記のコマンドを実行してください。
$ conda activate blast $ conda install pandas jupyter plotly ## pandas, jupyter, plotly 全てインストールされます。
y を選択して、完了してください。(*パッケージの依存関係等の問題で、仮想環境にトラブルが生じることがあります。その場合は、新な仮想環境を用意して、インストールしてください。今回利用するパッケージ同士の場合は、問題ないと思います。)
以下のコマンドを実行すれば、Jupyter notebook が起動します。
$ jupyter notebook
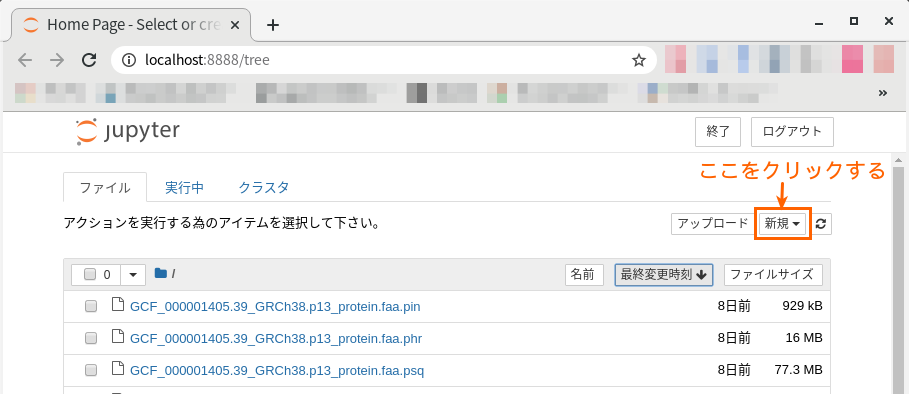
このようなブラウザが立ち上がれば、問題ありません。
「新規」という箇所をクリックして、jupyter notebookを作成し分析を開始しますが、今回はここまでにしたいと思います。続きは、以下の記事を参照してくださいね。
コメント